








DOI: https://doi.org/10.37349/eds.2024.00079
DOI: https://doi.org/10.37349/eds.2025.100896
Aim:
The biorecognition unit of an electrochemical biosensor requires molecules that are immobilised to serve as a bridge between the recognition unit and the transducing surface. Unique materials that enhance immobilisation of biorecognition molecules and improve electrochemical signal transduction are important in overcoming challenges based on the sensitivity of biosensors. In this regard, the electrochemical properties (EPs) of hydroxyapatite (HAp) material for the direct immobilisation of cells was investigated.
Methods:
Snail shell HAp (SHAp) material was synthesised from Achatina achatina snail shells and phosphate-containing solutions. The SHAp material was characterised using X-ray diffractometry (XRD), Fourier transform infrared (FTIR) spectroscopy, and Raman spectroscopy to determine the structural configuration, after which it was blended with a conductive polymer [poly(3,4-ethylenedioxythiophene): poly-4-styrene sulfonate (PEDOT: PSS)] to improve the electrochemical responses. The SHAp/PEDOT: PSS blend was used to modify a screen-printed carbon electrode (SPCE) by drop-casting, followed by seeding of pheochromocytoma (PC 12) and human embryonic kidney (HEK)-293T cells on the modified SPCE to record the EP using cyclic voltammetry (CV) and electrochemical impedance spectroscopy (EIS). Red blood cells (RBCs) were used as a control.
Results:
The CV analysis showed lower peak currents for HEK 293T (50 µA) and PC 12 (120 µA) compared to the RBC (230 µA). Also, the EIS showed impedance values of 0.70 for HEK 293T, 0.62 for PC 12, and 0.52 mΩ for RBC. The findings indicate that SHAp/PEDOT: PSS enables the differentiation of cell proliferation signals through voltammetric and impedimetric measurements.
Conclusions:
The unique current and impedance differences among the cells could serve as potential markers for rapid cell detection.
Aim:
The biorecognition unit of an electrochemical biosensor requires molecules that are immobilised to serve as a bridge between the recognition unit and the transducing surface. Unique materials that enhance immobilisation of biorecognition molecules and improve electrochemical signal transduction are important in overcoming challenges based on the sensitivity of biosensors. In this regard, the electrochemical properties (EPs) of hydroxyapatite (HAp) material for the direct immobilisation of cells was investigated.
Methods:
Snail shell HAp (SHAp) material was synthesised from Achatina achatina snail shells and phosphate-containing solutions. The SHAp material was characterised using X-ray diffractometry (XRD), Fourier transform infrared (FTIR) spectroscopy, and Raman spectroscopy to determine the structural configuration, after which it was blended with a conductive polymer [poly(3,4-ethylenedioxythiophene): poly-4-styrene sulfonate (PEDOT: PSS)] to improve the electrochemical responses. The SHAp/PEDOT: PSS blend was used to modify a screen-printed carbon electrode (SPCE) by drop-casting, followed by seeding of pheochromocytoma (PC 12) and human embryonic kidney (HEK)-293T cells on the modified SPCE to record the EP using cyclic voltammetry (CV) and electrochemical impedance spectroscopy (EIS). Red blood cells (RBCs) were used as a control.
Results:
The CV analysis showed lower peak currents for HEK 293T (50 µA) and PC 12 (120 µA) compared to the RBC (230 µA). Also, the EIS showed impedance values of 0.70 for HEK 293T, 0.62 for PC 12, and 0.52 mΩ for RBC. The findings indicate that SHAp/PEDOT: PSS enables the differentiation of cell proliferation signals through voltammetric and impedimetric measurements.
Conclusions:
The unique current and impedance differences among the cells could serve as potential markers for rapid cell detection.
DOI: https://doi.org/10.37349/eds.2023.00021
Green propolis is collected by Apis mellifera from the flowers and buds of Baccharis dracunculifolia. It has several chemical compounds that confer anti-inflammatory, antimicrobial, healing, and antioxidant biological activities. To report a series of clinical cases in the treatment of oral mucositis (OM) in patients with cancer undergoing radiotherapy in the head and neck region. Rapid treatment of OM means restoring quality of life to patients and lowering the cost of cancer treatment for public health. There male patients with oral carcinoma undergoing radiotherapy treatment were followed between August 2018 and April 2019. The patients presented themselves to the clinics in the Faculty of Dentistry of Federal University of Minas Gerais with erythematous and ulcerated coalescing lesions with purulent fibrin pseudomembranes in the oral mucosa, classified as grade IV OM according to the World Health Organization. The patients complained about the inability to eat, drink, and speak, which caused the radiotherapy interruption. After completing the clinical forms, anamnesis, and proper oral hygiene of each patient, a mucoadherent gel containing 5% propolis was prescribed for daily use, with a 3 time-a-day application every 8 h. After 7 days of use, there was an 80% lesion reduction, with total remission after 15 days of its application. The rapid response with total remission of lesions seems to be related to the chemical composition of propolis. Clinical and cellphone monitoring of patients, weekly and daily, respectively, were essential for successful treatment. The patients were monitored for one year, being encouraged to make constant use of the gel to control hyposalivation caused by changes in the salivary glands during radiotherapy.
Green propolis is collected by Apis mellifera from the flowers and buds of Baccharis dracunculifolia. It has several chemical compounds that confer anti-inflammatory, antimicrobial, healing, and antioxidant biological activities. To report a series of clinical cases in the treatment of oral mucositis (OM) in patients with cancer undergoing radiotherapy in the head and neck region. Rapid treatment of OM means restoring quality of life to patients and lowering the cost of cancer treatment for public health. There male patients with oral carcinoma undergoing radiotherapy treatment were followed between August 2018 and April 2019. The patients presented themselves to the clinics in the Faculty of Dentistry of Federal University of Minas Gerais with erythematous and ulcerated coalescing lesions with purulent fibrin pseudomembranes in the oral mucosa, classified as grade IV OM according to the World Health Organization. The patients complained about the inability to eat, drink, and speak, which caused the radiotherapy interruption. After completing the clinical forms, anamnesis, and proper oral hygiene of each patient, a mucoadherent gel containing 5% propolis was prescribed for daily use, with a 3 time-a-day application every 8 h. After 7 days of use, there was an 80% lesion reduction, with total remission after 15 days of its application. The rapid response with total remission of lesions seems to be related to the chemical composition of propolis. Clinical and cellphone monitoring of patients, weekly and daily, respectively, were essential for successful treatment. The patients were monitored for one year, being encouraged to make constant use of the gel to control hyposalivation caused by changes in the salivary glands during radiotherapy.
DOI: https://doi.org/10.37349/eds.2023.00022
Aim:
Modification of the C-terminus of a peptide to improve its properties, particularly after constructing the peptide chain, has great promise in the development of peptide therapeutics. This study discusses the development of a late-stage diversification method for synthesizing peptide acids and amides from hydrazides which can serve as a common precursor.
Methods:
Peptide hydrazides were synthesized solely by using conventional solid-phase peptide synthesis (SPPS). Hydrazides were subjected to oxidation by potassium peroxymonosulfate (Oxone) to afford carboxylic acids. Azidation of hydrazides using sodium nitrite (NaNO2) under acidic conditions, followed by the addition of β-mercaptoethanol (BME), could also be used to generate carboxylic acids. For the preparation of peptide amides, azides that can be prepared from hydrazides were reacted with ammonium acetate (NH4OAc) or tris(2-carboxyethyl)phosphine (TCEP)∙hydrochloride (HCl) to develop the products through ammonolysis or a Staudinger reaction, which produces iminophosphorane from an azide and a phosphine. The antimicrobial activity of modelin-5 derivatives synthesized from the corresponding hydrazides was evaluated by the colony count of Escherichia coli (E. coli) after treatment with the peptides.
Results:
Oxone oxidation yielded the corresponding acids rapidly although oxidation-prone amino acids were incompatible. Azidation and subsequent treatment with BME afforded peptide acids an acceptable yield even in sequences containing amino acids that are prone to oxidation. Both methods for conversion of hydrazides to amides were found to afford the desired products in good yield and compatibility. The conditions that were developed were adapted to the synthesis of modelin-5 derivatives from the corresponding hydrazides, yielding late-stage production of the desired peptides. The amides of the resulting peptide showed more potent activity against E. coli than the acid form, and the most potent activity was observed from the hydrazide.
Conclusions:
The developed protocols allow hydrazides to be converted to acids or amides, enabling late-stage diversification of peptide C-terminal residues.
Aim:
Modification of the C-terminus of a peptide to improve its properties, particularly after constructing the peptide chain, has great promise in the development of peptide therapeutics. This study discusses the development of a late-stage diversification method for synthesizing peptide acids and amides from hydrazides which can serve as a common precursor.
Methods:
Peptide hydrazides were synthesized solely by using conventional solid-phase peptide synthesis (SPPS). Hydrazides were subjected to oxidation by potassium peroxymonosulfate (Oxone) to afford carboxylic acids. Azidation of hydrazides using sodium nitrite (NaNO2) under acidic conditions, followed by the addition of β-mercaptoethanol (BME), could also be used to generate carboxylic acids. For the preparation of peptide amides, azides that can be prepared from hydrazides were reacted with ammonium acetate (NH4OAc) or tris(2-carboxyethyl)phosphine (TCEP)∙hydrochloride (HCl) to develop the products through ammonolysis or a Staudinger reaction, which produces iminophosphorane from an azide and a phosphine. The antimicrobial activity of modelin-5 derivatives synthesized from the corresponding hydrazides was evaluated by the colony count of Escherichia coli (E. coli) after treatment with the peptides.
Results:
Oxone oxidation yielded the corresponding acids rapidly although oxidation-prone amino acids were incompatible. Azidation and subsequent treatment with BME afforded peptide acids an acceptable yield even in sequences containing amino acids that are prone to oxidation. Both methods for conversion of hydrazides to amides were found to afford the desired products in good yield and compatibility. The conditions that were developed were adapted to the synthesis of modelin-5 derivatives from the corresponding hydrazides, yielding late-stage production of the desired peptides. The amides of the resulting peptide showed more potent activity against E. coli than the acid form, and the most potent activity was observed from the hydrazide.
Conclusions:
The developed protocols allow hydrazides to be converted to acids or amides, enabling late-stage diversification of peptide C-terminal residues.
DOI: https://doi.org/10.37349/eds.2023.00023
This article belongs to the special issue Bioactive Peptides discovery and development
The outbreak of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection in December 2019 quickly escalated to pandemic levels and had a severe impact on public health. There are 761 million confirmed coronavirus disease 2019 (COVID-19) cases, with over 6.88 million deaths worldwide till March 2023. Severe cases of the disease caused critical respiratory failure followed by multiorgan involvement. Clinical escalation of COVID-19 has been correlated with markedly increased plasma inflammatory markers [e.g., C-reactive protein (CRP)] and pro-inflammatory cytokine levels [e.g., interleukin (IL)-6, tumor necrosis factor-α (TNF-α)]. Therapeutic options have mostly utilized corticosteroids, antivirals (e.g., remdesivir), and monoclonal antibody-based immunomodulation (e.g., tocilizumab). These existing treatments have adverse side effects, inadequate efficacy, and limitations in administering to patients with comorbidities and other underlying diseases. Monoclonal antibody-based therapies and some of the antivirals are very costly. Many phytochemicals have previously reported anti-inflammatory, antiviral, and antioxidant properties. Studying the effectiveness of such phytochemicals against COVID-19 and identifying new plant-derived molecules with antiviral properties have been a focus since the SARS-CoV-2 outbreak. This review article has documented in vitro, in vivo, and clinical studies encompassing 28 different phytochemicals belonging to various chemical groups (e.g., polyphenols, alkaloids, terpenes) that show anti-COVID-19 activity. These findings suggest that multiple phytochemicals can interfere with virus entry and replication inside the host cell. Many of them can protect from cytokine storm by acting on intracellular signalling pathways in addition to inhibiting virus multiplication. Phytochemicals may prove useful in alleviating post-COVID complications associated with kidney injury, and central nervous system complications, as well. Plant-derived compounds are usually cheaper and have fewer side effects. But, developing new formulations with better absorption and bioavailability remains a priority. This review informs the readers of the current status and indicates the ongoing research in this highly relevant field.
The outbreak of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection in December 2019 quickly escalated to pandemic levels and had a severe impact on public health. There are 761 million confirmed coronavirus disease 2019 (COVID-19) cases, with over 6.88 million deaths worldwide till March 2023. Severe cases of the disease caused critical respiratory failure followed by multiorgan involvement. Clinical escalation of COVID-19 has been correlated with markedly increased plasma inflammatory markers [e.g., C-reactive protein (CRP)] and pro-inflammatory cytokine levels [e.g., interleukin (IL)-6, tumor necrosis factor-α (TNF-α)]. Therapeutic options have mostly utilized corticosteroids, antivirals (e.g., remdesivir), and monoclonal antibody-based immunomodulation (e.g., tocilizumab). These existing treatments have adverse side effects, inadequate efficacy, and limitations in administering to patients with comorbidities and other underlying diseases. Monoclonal antibody-based therapies and some of the antivirals are very costly. Many phytochemicals have previously reported anti-inflammatory, antiviral, and antioxidant properties. Studying the effectiveness of such phytochemicals against COVID-19 and identifying new plant-derived molecules with antiviral properties have been a focus since the SARS-CoV-2 outbreak. This review article has documented in vitro, in vivo, and clinical studies encompassing 28 different phytochemicals belonging to various chemical groups (e.g., polyphenols, alkaloids, terpenes) that show anti-COVID-19 activity. These findings suggest that multiple phytochemicals can interfere with virus entry and replication inside the host cell. Many of them can protect from cytokine storm by acting on intracellular signalling pathways in addition to inhibiting virus multiplication. Phytochemicals may prove useful in alleviating post-COVID complications associated with kidney injury, and central nervous system complications, as well. Plant-derived compounds are usually cheaper and have fewer side effects. But, developing new formulations with better absorption and bioavailability remains a priority. This review informs the readers of the current status and indicates the ongoing research in this highly relevant field.
DOI: https://doi.org/10.37349/eds.2023.00024
This article belongs to the special issue Exploring Potential Drugs from Natural Products
Protein therapeutics are extensively used in the treatment of autoimmune diseases, but a subset of patients appears to be refractory to these treatments, mainly due to the development of an immune response to the drug. A better understanding of the mechanism underlying the therapeutic drug’s failure becomes fundamental for the development of new and more effective treatments. Unfortunately, there are few cases where the exact mechanisms through which drugs bypass immunological tolerance and provoke immunogenicity have been studied. In this context, peptide epitope identification gained increasing importance in investigating the molecular mechanism of therapeutic drug’s immune responses. Despite peptide identification and use to monitor anti-drug antibody (ADA) profiles is a promising research field, their use is far away from a wide application both at the research and at the commercial level. Herein it is reported a compilation of studies in which peptides are directly involved in anti-drug immune responses, becoming the molecular key step for a better understanding of refractory reactions in therapeutic drugs. An overview on T-cell and B-cell peptide recognition is given, showing the growing potential and advantages of peptides when used in the field of refractoriness to drugs. This review includes studies describing antigenic peptides that enable enhanced ADA detection directly in patients’ sera, as well as the proof of concept that asses the use of peptides instead of proteins, to facilitate the identification of neutralizing ADA.
Protein therapeutics are extensively used in the treatment of autoimmune diseases, but a subset of patients appears to be refractory to these treatments, mainly due to the development of an immune response to the drug. A better understanding of the mechanism underlying the therapeutic drug’s failure becomes fundamental for the development of new and more effective treatments. Unfortunately, there are few cases where the exact mechanisms through which drugs bypass immunological tolerance and provoke immunogenicity have been studied. In this context, peptide epitope identification gained increasing importance in investigating the molecular mechanism of therapeutic drug’s immune responses. Despite peptide identification and use to monitor anti-drug antibody (ADA) profiles is a promising research field, their use is far away from a wide application both at the research and at the commercial level. Herein it is reported a compilation of studies in which peptides are directly involved in anti-drug immune responses, becoming the molecular key step for a better understanding of refractory reactions in therapeutic drugs. An overview on T-cell and B-cell peptide recognition is given, showing the growing potential and advantages of peptides when used in the field of refractoriness to drugs. This review includes studies describing antigenic peptides that enable enhanced ADA detection directly in patients’ sera, as well as the proof of concept that asses the use of peptides instead of proteins, to facilitate the identification of neutralizing ADA.
DOI: https://doi.org/10.37349/eds.2023.00025
This article belongs to the special issue Bioactive Peptides discovery and development
Aim:
Solubility prediction is an essential factor in rational drug design and many models have been developed with machine learning (ML) methods to enhance the predictive ability. However, most of the ML models are hard to interpret which limits the insights they can give in the lead optimization process. Here, an approach to construct and interpret solubility models with a combination of physicochemical properties and ML algorithms is presented.
Methods:
The models were trained, optimized, and tested in a dataset containing 12,983 compounds from two public datasets and further evaluated in two external test sets. More importantly, the SHapley Additive exPlanations (SHAP) and heat map coloring approaches were used to explain the predictive models and assess their suitability to guide compound optimization.
Results:
Among the different ML methods, random forest (RF) models obtain the best performance in the different test sets. From the interpretability perspective, fragment-based coloring offers a more robust interpretation than atom-based coloring and that normalizing the values further improves it.
Conclusions:
Overall, for certain applications simple ML algorithms such as RF work well and can outperform more complex methods and that combining them with fragment-coloring can offer guidance for chemists to modify the structure with a desired property. This interpretation strategy is publicly available at https://github.com/Pharmacelera/predictive-model-coloring and could be further applied in other property predictions to improve the interpretability of ML models.
Aim:
Solubility prediction is an essential factor in rational drug design and many models have been developed with machine learning (ML) methods to enhance the predictive ability. However, most of the ML models are hard to interpret which limits the insights they can give in the lead optimization process. Here, an approach to construct and interpret solubility models with a combination of physicochemical properties and ML algorithms is presented.
Methods:
The models were trained, optimized, and tested in a dataset containing 12,983 compounds from two public datasets and further evaluated in two external test sets. More importantly, the SHapley Additive exPlanations (SHAP) and heat map coloring approaches were used to explain the predictive models and assess their suitability to guide compound optimization.
Results:
Among the different ML methods, random forest (RF) models obtain the best performance in the different test sets. From the interpretability perspective, fragment-based coloring offers a more robust interpretation than atom-based coloring and that normalizing the values further improves it.
Conclusions:
Overall, for certain applications simple ML algorithms such as RF work well and can outperform more complex methods and that combining them with fragment-coloring can offer guidance for chemists to modify the structure with a desired property. This interpretation strategy is publicly available at https://github.com/Pharmacelera/predictive-model-coloring and could be further applied in other property predictions to improve the interpretability of ML models.
DOI: https://doi.org/10.37349/eds.2023.00026
This article belongs to the special issue Machine Learning for Drug Science
Aim:
Harzianoic acids A and B (Hz-A/B) are two rare cyclobutene-containing sesquiterpenes isolated from a marine strain of the sponge-associated fungus Trichoderma harzianum. They display anticancer and antiviral effects, reducing the entry of hepatitis C virus (HCV) into hepatocarcinoma cells. The large extracellular loop (LEL) of the tetraspanin protein CD81 represents a molecular target for both Hz-A and Hz-B.
Methods:
The interaction of Hz-A/B with CD81 has been modeled, using structures of the cholesterol-bound full-length protein and a truncated protein corresponding to the LEL portion. The models mimicked the closed and open conformations of the LEL.
Results:
The best ligand Hz-B can form stable complexes with the open LEL structure, whereas binding to the closed form is drastically reduced. Key H-bonds between the acid groups of Hz-B and the CD81-LEL domain stabilize the ligand-protein complex. A comparison of the interaction with the homologous tetraspanin CD9, which also presents a dynamic open/closed equilibrium, underlined the marked selectivity of Hz-A/B for CD81 over CD9. The cyclobutane-containing monoterpene grandisol, an insect pheromone, has been identified as a fragment that could be modulated to improve its modest interaction with CD81-LEL.
Conclusions:
The modeling docking analysis suggests that Hz-B is a robust CD81 binder, better interacting with the LEL portion of CD81 compared to CD9-LEL. The docking study paves the way to the design of small molecules targeting CD81. The study has implications for a better understanding of CD81 binding properties and the regulation of its activities.
Aim:
Harzianoic acids A and B (Hz-A/B) are two rare cyclobutene-containing sesquiterpenes isolated from a marine strain of the sponge-associated fungus Trichoderma harzianum. They display anticancer and antiviral effects, reducing the entry of hepatitis C virus (HCV) into hepatocarcinoma cells. The large extracellular loop (LEL) of the tetraspanin protein CD81 represents a molecular target for both Hz-A and Hz-B.
Methods:
The interaction of Hz-A/B with CD81 has been modeled, using structures of the cholesterol-bound full-length protein and a truncated protein corresponding to the LEL portion. The models mimicked the closed and open conformations of the LEL.
Results:
The best ligand Hz-B can form stable complexes with the open LEL structure, whereas binding to the closed form is drastically reduced. Key H-bonds between the acid groups of Hz-B and the CD81-LEL domain stabilize the ligand-protein complex. A comparison of the interaction with the homologous tetraspanin CD9, which also presents a dynamic open/closed equilibrium, underlined the marked selectivity of Hz-A/B for CD81 over CD9. The cyclobutane-containing monoterpene grandisol, an insect pheromone, has been identified as a fragment that could be modulated to improve its modest interaction with CD81-LEL.
Conclusions:
The modeling docking analysis suggests that Hz-B is a robust CD81 binder, better interacting with the LEL portion of CD81 compared to CD9-LEL. The docking study paves the way to the design of small molecules targeting CD81. The study has implications for a better understanding of CD81 binding properties and the regulation of its activities.
DOI: https://doi.org/10.37349/eds.2023.00027
Aim:
This study discloses the synthesis and the antimicrobial and anticancer activities of four molecules of structural basis saccharin-thiadiazolyl (4), saccharin-pyridyl (6, 8), and tetrazole-thiadiazolyl (11).
Methods:
Antimicrobial properties of the molecules were evaluated by the well-diffusion method, against Gram-positive bacteria [Staphylococcus aureus American Type Culture Collection (ATCC) 25923, Staphylococcus epidermidis ATCC 12228, Mycobacterium smegmatis ATCC 607], Gram-negative bacteria (Pseudomonas aeruginosa ATCC 27853) and yeast (Saccharomyces cerevisiae ATCC 2601 and Candida albicans ATCC 10231) strains. The anticancer activity of the compounds was assessed through i) proliferation assays for HCT116, MCF-7, and A375 human cell lines [cells were treated with serial dilutions of compounds and the effect on cell propagation was evaluated by sulforhodamine B (SRB) assay]; ii) antiproliferative and cytotoxic assays for glioma-type cell lines A172 (glioblastoma), U87 (brain-likely glioblastoma), and H4 (neuroglioma; cells were treated with diverse concentrations and the cell viability was assessed using a modified Alamar blue® assay).
Results:
Compound 11 exhibited significant inhibitory activity against S. aureus and S. epidermidis, with the further molecules demonstrating some inhibitory potential against all the tested Gram-positive, Gram-negative, and yeast strains. Similarly, derivative 11 showed an interesting antiproliferative activity against human colon adenocarcinoma (HCT116), human breast adenocarcinoma (MCF-7), and melanoma (A375) cells, with 50% growth inhibition (GI50) values varying from 3.55 µmol/L to 11.5 µmol/L, in the same order of magnitude of those shown by etoposide. Treatment of brain-like glioblastoma cells (U87) with 11, at the concentration of 100 µg/mL, induced a decrease in cell viability by 50% after 48 h and 72 h. Besides, results attained for A172 cells have shown that compound 11 only induces a significant decrease in cell viability upon treatment at 100 µg/mL for 72 h. A divergent observation was recorded for H4 cells, where the treatment with derivative 11 had promoted a significant decrease in cell viability (< 40–60%), even at concentrations as low as 0.39 µg/mL, after 24 h.
Conclusions:
This investigation reveals the potential of distinct azole-based conjugates, in particular the tetrazole-thiadiazolyl (11) derivative, as scaffolds worth further investigations, in the frame of antimicrobial and antineoplastic chemotherapy.
Aim:
This study discloses the synthesis and the antimicrobial and anticancer activities of four molecules of structural basis saccharin-thiadiazolyl (4), saccharin-pyridyl (6, 8), and tetrazole-thiadiazolyl (11).
Methods:
Antimicrobial properties of the molecules were evaluated by the well-diffusion method, against Gram-positive bacteria [Staphylococcus aureus American Type Culture Collection (ATCC) 25923, Staphylococcus epidermidis ATCC 12228, Mycobacterium smegmatis ATCC 607], Gram-negative bacteria (Pseudomonas aeruginosa ATCC 27853) and yeast (Saccharomyces cerevisiae ATCC 2601 and Candida albicans ATCC 10231) strains. The anticancer activity of the compounds was assessed through i) proliferation assays for HCT116, MCF-7, and A375 human cell lines [cells were treated with serial dilutions of compounds and the effect on cell propagation was evaluated by sulforhodamine B (SRB) assay]; ii) antiproliferative and cytotoxic assays for glioma-type cell lines A172 (glioblastoma), U87 (brain-likely glioblastoma), and H4 (neuroglioma; cells were treated with diverse concentrations and the cell viability was assessed using a modified Alamar blue® assay).
Results:
Compound 11 exhibited significant inhibitory activity against S. aureus and S. epidermidis, with the further molecules demonstrating some inhibitory potential against all the tested Gram-positive, Gram-negative, and yeast strains. Similarly, derivative 11 showed an interesting antiproliferative activity against human colon adenocarcinoma (HCT116), human breast adenocarcinoma (MCF-7), and melanoma (A375) cells, with 50% growth inhibition (GI50) values varying from 3.55 µmol/L to 11.5 µmol/L, in the same order of magnitude of those shown by etoposide. Treatment of brain-like glioblastoma cells (U87) with 11, at the concentration of 100 µg/mL, induced a decrease in cell viability by 50% after 48 h and 72 h. Besides, results attained for A172 cells have shown that compound 11 only induces a significant decrease in cell viability upon treatment at 100 µg/mL for 72 h. A divergent observation was recorded for H4 cells, where the treatment with derivative 11 had promoted a significant decrease in cell viability (< 40–60%), even at concentrations as low as 0.39 µg/mL, after 24 h.
Conclusions:
This investigation reveals the potential of distinct azole-based conjugates, in particular the tetrazole-thiadiazolyl (11) derivative, as scaffolds worth further investigations, in the frame of antimicrobial and antineoplastic chemotherapy.
DOI: https://doi.org/10.37349/eds.2023.00028
DOI: https://doi.org/10.37349/eds.2022.00001
Aim:
The aim of this research work was to develop a validated reversed-phase (RP)-high-performance liquid chromatography (HPLC) method for simultaneous estimation of oxytetracycline (OXY) and polymixin B (PMB) in fixed-dose combination.
Methods:
The HPLC assay method was validated on X-Bridge C18 [250 mm × 4.6 mm intradermal (i.d.), 5 μm], mobile phase consisting of aotearoa co-incidence network (ACN):water containing 0.5% (v/v) orthophosphoric acid (pH 3.5) in the ratio of 80:20 respectively. The flow rate was set at 0.9 mL/min and the column was maintained at room temperature. The RP-HPLC method was validated in terms of the calibration curve (CC), linearity and range, limit of detection (LOD), and limit of quantitation (LOQ), precision, robustness, and accuracy.
Results:
The method was found to be linear with a concentration range of 5–25 μg/mL. Precision results showed the developed method was found to be precise with a relative standard deviation [RSD (%)] value < 2. Accuracy showed acceptable recovery of prepared concentrations as per International Conference on Harmonization (ICH) guidelines. Moreover, the developed method was found to be robust and rugged, as per specified ranges. The assay of these two drugs in marketed formulation, i.e., Terramycin® Ointment showed satisfactory recovery, as per ICH guidelines. The results proved that the method can be used for the routine-based estimation of OXY and PMB.
Conclusions:
Linear CC were obtained with a correlation coefficient (R2 > 0.99) with acceptable results of accuracy and precision.
Ultraviolet visible and HPLC method development
Aim:
The aim of this research work was to develop a validated reversed-phase (RP)-high-performance liquid chromatography (HPLC) method for simultaneous estimation of oxytetracycline (OXY) and polymixin B (PMB) in fixed-dose combination.
Methods:
The HPLC assay method was validated on X-Bridge C18 [250 mm × 4.6 mm intradermal (i.d.), 5 μm], mobile phase consisting of aotearoa co-incidence network (ACN):water containing 0.5% (v/v) orthophosphoric acid (pH 3.5) in the ratio of 80:20 respectively. The flow rate was set at 0.9 mL/min and the column was maintained at room temperature. The RP-HPLC method was validated in terms of the calibration curve (CC), linearity and range, limit of detection (LOD), and limit of quantitation (LOQ), precision, robustness, and accuracy.
Results:
The method was found to be linear with a concentration range of 5–25 μg/mL. Precision results showed the developed method was found to be precise with a relative standard deviation [RSD (%)] value < 2. Accuracy showed acceptable recovery of prepared concentrations as per International Conference on Harmonization (ICH) guidelines. Moreover, the developed method was found to be robust and rugged, as per specified ranges. The assay of these two drugs in marketed formulation, i.e., Terramycin® Ointment showed satisfactory recovery, as per ICH guidelines. The results proved that the method can be used for the routine-based estimation of OXY and PMB.
Conclusions:
Linear CC were obtained with a correlation coefficient (R2 > 0.99) with acceptable results of accuracy and precision.
Ultraviolet visible and HPLC method development
DOI: https://doi.org/10.37349/eds.2023.00002
Aim:
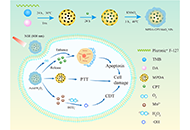
The development of a collaborative strategy with improved efficacy holds great promise in tumor treatment. This study aims to develop an effective collaborative strategy based on functionalized mesoporous polydopamine (MPDA) nanocomposites for killing tumor cells.
Methods:
MPDA nanoparticles were synthesized and functionalized with camptothecin (CPT) payload and manganese dioxide (MnO2) coating to construct MPDA-CPT-MnO2 nanocomposites.
Results:
When uptaken by tumor cells, the nanocomposites can degrade to produce O2, release CPT, and generate manganese (Mn2+) under the stimulation of hydrogen peroxide (H2O2) and acid. The released CPT and Mn2+ can act as chemotherapeutic drug and Fenton-like agent, respectively. Abundant reactive oxygen species (ROS) are generated in 4T1 tumor cells through an Mn2+-mediated Fenton-like reaction. After that, the generated Mn4+ can react with glutathione (GSH) through redox reaction to produce Mn2+ and deplete GSH, disrupting the reducing capacity and benefiting the production of ROS in tumor cells. Under laser irradiation, the nanocomposites can generate hyperthermia to promote the production of ROS.
Conclusions:
The developed MPDA-CPT-MnO2 nanocomposites can kill tumor cells through collaborative chemo/photothermal/chemodynamic therapy (CDT).
Aim:
The development of a collaborative strategy with improved efficacy holds great promise in tumor treatment. This study aims to develop an effective collaborative strategy based on functionalized mesoporous polydopamine (MPDA) nanocomposites for killing tumor cells.
Methods:
MPDA nanoparticles were synthesized and functionalized with camptothecin (CPT) payload and manganese dioxide (MnO2) coating to construct MPDA-CPT-MnO2 nanocomposites.
Results:
When uptaken by tumor cells, the nanocomposites can degrade to produce O2, release CPT, and generate manganese (Mn2+) under the stimulation of hydrogen peroxide (H2O2) and acid. The released CPT and Mn2+ can act as chemotherapeutic drug and Fenton-like agent, respectively. Abundant reactive oxygen species (ROS) are generated in 4T1 tumor cells through an Mn2+-mediated Fenton-like reaction. After that, the generated Mn4+ can react with glutathione (GSH) through redox reaction to produce Mn2+ and deplete GSH, disrupting the reducing capacity and benefiting the production of ROS in tumor cells. Under laser irradiation, the nanocomposites can generate hyperthermia to promote the production of ROS.
Conclusions:
The developed MPDA-CPT-MnO2 nanocomposites can kill tumor cells through collaborative chemo/photothermal/chemodynamic therapy (CDT).
DOI: https://doi.org/10.37349/eds.2023.00003
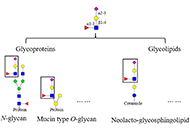
It is widely acknowledged that sialyl Lewis X (sLeX), the composition and linkage of which are N-acetylneuraminic acid (Neu5Ac) α2-3 galactose (Gal) β1-4 [fucose (Fuc) α1-3] N-acetylglucosamine, is usually attached to the cell surface. It presents as a terminal structure on either glycoproteins or glycolipids and has been demonstrated to be related to various biological processes, such as fertilization and selectin binding. Due to the vital role of sLeX, its synthesis as well as its determination approaches have attracted considerable attention from many researchers. In this review, the focus is sLeX on glycoproteins. The biological importance of sLeX in fertilization and development, immunity, cancers, and other aspects will be first introduced. Then the chemical and enzymatic synthesis of sLeX including the contributions from more than 15 international research groups will be described, followed by a brief view of the sLeX detection focusing on monosaccharides and linkages. This review is valuable for those readers who are interested in the chemistry and biology of sLeX.
It is widely acknowledged that sialyl Lewis X (sLeX), the composition and linkage of which are N-acetylneuraminic acid (Neu5Ac) α2-3 galactose (Gal) β1-4 [fucose (Fuc) α1-3] N-acetylglucosamine, is usually attached to the cell surface. It presents as a terminal structure on either glycoproteins or glycolipids and has been demonstrated to be related to various biological processes, such as fertilization and selectin binding. Due to the vital role of sLeX, its synthesis as well as its determination approaches have attracted considerable attention from many researchers. In this review, the focus is sLeX on glycoproteins. The biological importance of sLeX in fertilization and development, immunity, cancers, and other aspects will be first introduced. Then the chemical and enzymatic synthesis of sLeX including the contributions from more than 15 international research groups will be described, followed by a brief view of the sLeX detection focusing on monosaccharides and linkages. This review is valuable for those readers who are interested in the chemistry and biology of sLeX.
DOI: https://doi.org/10.37349/eds.2023.00004
This article belongs to the special issue Bioactive Peptides discovery and development
Aim:
Conventional techniques to share and archive spinal imaging data raise issues with trust and security, with novel approaches being more greatly considered. Ethereum smart contracts present one such novel approach. Ethereum is an open-source platform that allows for the use of smart contracts. Smart contracts are packages of code that are self-executing and reside in the Ethereum state, defining conditions for programmed transactions. Though powerful, limited attempts have been made to showcase the clinical utility of such technologies, especially in the pre- and post-operative imaging arenas. Herein, we therefore aim to propose a proof-of-concept smart contract that stores intraoperative three-dimensional (3D) augmented reality surgical navigation (ARSN) data and was tested on a private, proof-of-authority network. To the author’s best knowledge, the present study represents a first-use case of the InterPlanetary File Storage protocol for storing and retrieving spine imaging smart contracts.
Methods:
The content identifier hashes were stored inside the smart contracts while the interplanetary file system (IPFS) was used to efficiently store the image files. Insertion was achieved with four storage mappings, one for each of the following: fictitious patient data, specific diagnosis, patient identity document (ID), and Gertzbein grade. Inserted patient observations were then queried with wildcards. Insertion and retrieval times for different record volumes were collected.
Results:
It took 276 milliseconds to insert 50 records and 713 milliseconds to insert 350 records. Inserting 50 records required 934 Megabyte (MB) of memory per insertion with patient data and imaging, while inserting 350 records required almost the same amount of memory per insertion. In a database of 350 records, the retrieval function needs about 1,026 MB to query a record with all three fields left blank, but only 970 MB to obtain the same observation from a database of 50 records.
Conclusions:
The concept presented in this study exemplifies the clinical utility of smart contracts and off-chain data storage for efficient retrieval/insertion of ARSN data.
Aim:
Conventional techniques to share and archive spinal imaging data raise issues with trust and security, with novel approaches being more greatly considered. Ethereum smart contracts present one such novel approach. Ethereum is an open-source platform that allows for the use of smart contracts. Smart contracts are packages of code that are self-executing and reside in the Ethereum state, defining conditions for programmed transactions. Though powerful, limited attempts have been made to showcase the clinical utility of such technologies, especially in the pre- and post-operative imaging arenas. Herein, we therefore aim to propose a proof-of-concept smart contract that stores intraoperative three-dimensional (3D) augmented reality surgical navigation (ARSN) data and was tested on a private, proof-of-authority network. To the author’s best knowledge, the present study represents a first-use case of the InterPlanetary File Storage protocol for storing and retrieving spine imaging smart contracts.
Methods:
The content identifier hashes were stored inside the smart contracts while the interplanetary file system (IPFS) was used to efficiently store the image files. Insertion was achieved with four storage mappings, one for each of the following: fictitious patient data, specific diagnosis, patient identity document (ID), and Gertzbein grade. Inserted patient observations were then queried with wildcards. Insertion and retrieval times for different record volumes were collected.
Results:
It took 276 milliseconds to insert 50 records and 713 milliseconds to insert 350 records. Inserting 50 records required 934 Megabyte (MB) of memory per insertion with patient data and imaging, while inserting 350 records required almost the same amount of memory per insertion. In a database of 350 records, the retrieval function needs about 1,026 MB to query a record with all three fields left blank, but only 970 MB to obtain the same observation from a database of 50 records.
Conclusions:
The concept presented in this study exemplifies the clinical utility of smart contracts and off-chain data storage for efficient retrieval/insertion of ARSN data.
DOI: https://doi.org/10.37349/eds.2023.00005
Aim:
In the present study, the natural products levistolide A (LA) and periplogenin (PPG) were studied for their growth inhibitory effects on the development of gastric cancer cells in vitro and, more critically, in vivo, alone or in combination with the therapeutic medication 5-fluorouracil (5-FU).
Methods:
Methyl thiazolyl tetrazolium (MTT), also known as 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide assays were used for the cell viability study. Apoptosis was detected by western blot to detect the cleavage of caspase substrate poly (ADP-ribose) polymerase (PARP) and terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate (dUTP) nick-end labelling (TUNEL) assays. The nude mice bearing gastric cancer cells were used for the anti-cancer activity detection of LA and its combinational treatment effect with 5-FU.
Results:
The results in the present study shown that the two compounds were able to inhibit the viability of the cancer cells in a dose- and time-dependent manner by MTT method. They could trigger apoptosis when used alone, and more potently, in combination with 5-FU detected by TUNEL positivity and the cleavage of caspase substrate PARP. In nude mice bearing gastric cancer cells, injection (i.p.) of LA or PPG alone inhibited the growth of the cancer cells. The treatment using one of the compounds in combination with 5-FU inhibited the cancer cell growth at a higher level than the treatment by a compound alone.
Conclusions:
LA and PPG could inhibit the growth of the cancer cells, alone or in combination with 5-FU, in vitro and in vivo, suggesting that they are promising investigational drugs for therapeutic development.
Aim:
In the present study, the natural products levistolide A (LA) and periplogenin (PPG) were studied for their growth inhibitory effects on the development of gastric cancer cells in vitro and, more critically, in vivo, alone or in combination with the therapeutic medication 5-fluorouracil (5-FU).
Methods:
Methyl thiazolyl tetrazolium (MTT), also known as 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide assays were used for the cell viability study. Apoptosis was detected by western blot to detect the cleavage of caspase substrate poly (ADP-ribose) polymerase (PARP) and terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate (dUTP) nick-end labelling (TUNEL) assays. The nude mice bearing gastric cancer cells were used for the anti-cancer activity detection of LA and its combinational treatment effect with 5-FU.
Results:
The results in the present study shown that the two compounds were able to inhibit the viability of the cancer cells in a dose- and time-dependent manner by MTT method. They could trigger apoptosis when used alone, and more potently, in combination with 5-FU detected by TUNEL positivity and the cleavage of caspase substrate PARP. In nude mice bearing gastric cancer cells, injection (i.p.) of LA or PPG alone inhibited the growth of the cancer cells. The treatment using one of the compounds in combination with 5-FU inhibited the cancer cell growth at a higher level than the treatment by a compound alone.
Conclusions:
LA and PPG could inhibit the growth of the cancer cells, alone or in combination with 5-FU, in vitro and in vivo, suggesting that they are promising investigational drugs for therapeutic development.
DOI: https://doi.org/10.37349/eds.2023.00006
This article belongs to the special issue Exploring Potential Drugs from Natural Products
DOI: https://doi.org/10.37349/eds.2023.00007
This article belongs to the special issue Machine Learning for Drug Science
Malignant brain tumors are the leading cause of cancer-related death in children and remain a significant cause of morbidity and mortality throughout all demographics. Central nervous system (CNS) tumors are classically treated with surgical resection and radiotherapy in addition to adjuvant chemotherapy. However, the therapeutic efficacy of chemotherapeutic agents is limited due to the blood-brain barrier (BBB). Magnetic resonance guided focused ultrasound (MRgFUS) is a new and promising intervention for CNS tumors, which has shown success in preclinical trials. High-intensity focused ultrasound (HIFU) has the capacity to serve as a direct therapeutic agent in the form of thermoablation and mechanical destruction of the tumor. Low-intensity focused ultrasound (LIFU) has been shown to disrupt the BBB and enhance the uptake of therapeutic agents in the brain and CNS. The authors present a review of MRgFUS in the treatment of CNS tumors. This treatment method has shown promising results in preclinical trials including minimal adverse effects, increased infiltration of the therapeutic agents into the CNS, decreased tumor progression, and improved survival rates.
Malignant brain tumors are the leading cause of cancer-related death in children and remain a significant cause of morbidity and mortality throughout all demographics. Central nervous system (CNS) tumors are classically treated with surgical resection and radiotherapy in addition to adjuvant chemotherapy. However, the therapeutic efficacy of chemotherapeutic agents is limited due to the blood-brain barrier (BBB). Magnetic resonance guided focused ultrasound (MRgFUS) is a new and promising intervention for CNS tumors, which has shown success in preclinical trials. High-intensity focused ultrasound (HIFU) has the capacity to serve as a direct therapeutic agent in the form of thermoablation and mechanical destruction of the tumor. Low-intensity focused ultrasound (LIFU) has been shown to disrupt the BBB and enhance the uptake of therapeutic agents in the brain and CNS. The authors present a review of MRgFUS in the treatment of CNS tumors. This treatment method has shown promising results in preclinical trials including minimal adverse effects, increased infiltration of the therapeutic agents into the CNS, decreased tumor progression, and improved survival rates.
DOI: https://doi.org/10.37349/eds.2023.00009
Bladder cancer (BC) is a complex disease with multiple clinical manifestations and treatment challenges, and current standard-of-care therapies remain limited and unfavorable. Theranostics, the integration of diagnostic and therapeutic technologies, has emerged as a promising strategy to address these challenges. The rapid development of nanomedicine has been a source of hope for the improvement of BC therapies and diagnostics by reducing side effects, enhancing tumor suppression, and overcoming drug resistance. Metal nanoparticles (NPs), inorganic NPs, polymer NPs, etc. have their respective advantages and show encouraging potential in the therapy of BC. In this review, we provide an overview on the state of the art in nanotechnology-based theranostics for BC, offering insights into the design and discovery of novel NPs for future BC management.
Bladder cancer (BC) is a complex disease with multiple clinical manifestations and treatment challenges, and current standard-of-care therapies remain limited and unfavorable. Theranostics, the integration of diagnostic and therapeutic technologies, has emerged as a promising strategy to address these challenges. The rapid development of nanomedicine has been a source of hope for the improvement of BC therapies and diagnostics by reducing side effects, enhancing tumor suppression, and overcoming drug resistance. Metal nanoparticles (NPs), inorganic NPs, polymer NPs, etc. have their respective advantages and show encouraging potential in the therapy of BC. In this review, we provide an overview on the state of the art in nanotechnology-based theranostics for BC, offering insights into the design and discovery of novel NPs for future BC management.
DOI: https://doi.org/10.37349/eds.2023.00008
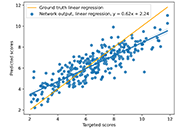
Aim:
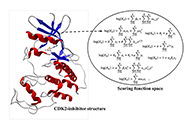
Drug discovery is a long process, often taking decades of research endeavors. It is still an active area of research in both academic and industrial sectors with efforts on reducing time and cost. Computational simulations like molecular docking enable fast exploration of large databases of compounds and extract the most promising molecule candidates for further in vitro and in vivo tests. Structure-based molecular docking is a complex process mixing both surface exploration and energy estimation to find the minimal free energy of binding corresponding to the best interaction location.
Methods:
Hereafter, heterogeneous graph score (HGScore), a new scoring function is proposed and is developed in the context of a protein-small compound-complex. Each complex is represented by a heterogeneous graph allowing to separate edges according to their class (inter- or intra-molecular). Then a heterogeneous graph convolutional network (HGCN) is used allowing the discrimination of the information according to the edge crossed. In the end, the model produces the affinity score of the complex.
Results:
HGScore has been tested on the comparative assessment of scoring functions (CASF) 2013 and 2016 benchmarks for scoring, ranking, and docking powers. It has achieved good performances by outperforming classical methods and being among the best artificial intelligence (AI) methods.
Conclusions:
Thus, HGScore brings a new way to represent protein-ligand interactions. Using a representation that involves classical graph neural networks (GNNs) and splitting the learning process regarding the edge type makes the proposed model to be the best adapted for future transfer learning on other (protein-DNA, protein-sugar, protein-protein, etc.) biological complexes.
Aim:
Drug discovery is a long process, often taking decades of research endeavors. It is still an active area of research in both academic and industrial sectors with efforts on reducing time and cost. Computational simulations like molecular docking enable fast exploration of large databases of compounds and extract the most promising molecule candidates for further in vitro and in vivo tests. Structure-based molecular docking is a complex process mixing both surface exploration and energy estimation to find the minimal free energy of binding corresponding to the best interaction location.
Methods:
Hereafter, heterogeneous graph score (HGScore), a new scoring function is proposed and is developed in the context of a protein-small compound-complex. Each complex is represented by a heterogeneous graph allowing to separate edges according to their class (inter- or intra-molecular). Then a heterogeneous graph convolutional network (HGCN) is used allowing the discrimination of the information according to the edge crossed. In the end, the model produces the affinity score of the complex.
Results:
HGScore has been tested on the comparative assessment of scoring functions (CASF) 2013 and 2016 benchmarks for scoring, ranking, and docking powers. It has achieved good performances by outperforming classical methods and being among the best artificial intelligence (AI) methods.
Conclusions:
Thus, HGScore brings a new way to represent protein-ligand interactions. Using a representation that involves classical graph neural networks (GNNs) and splitting the learning process regarding the edge type makes the proposed model to be the best adapted for future transfer learning on other (protein-DNA, protein-sugar, protein-protein, etc.) biological complexes.
DOI: https://doi.org/10.37349/eds.2023.00010
This article belongs to the special issue Machine Learning for Drug Science
