



DOI: https://doi.org/10.37349/ei.2024.00169
This review pretends to shed light on the immune processes occurring in the coronavirus disease 2019 (COVID-19) from a perspective based on the antigens size, lower or larger than 70 kDa. This cutoff size point explains the host type of immune response against the antigenic proteins of the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), which may lead to the development of the memory B cells or, conversely, the immune suppression, apoptosis, viral escape, and sepsis. Here, based on previous experimental work and the review of related literature, the following is proposed: antigens < 70 kDa can access the germinal center through the follicular conduits, where the activated B cells can present the processed antigen to specific naive CD4+ T cells that, in interaction with the major histocompatibility complex class II (MHC-II), trigger the immune response T helper type 2 (Th2). Conversely, antigens > 70 kDa cannot circulate through the narrow follicular conduits network and might be captured within the subcapsular sinus by the macrophages and dendritic follicular cells. Then, these cognate antigens are presented, via complement receptors, to the B cells that acquire and present them through the MHC-II to the specific naive CD4+ T cells, triggering the immune response Th1. The sustained infected cells lysis can overfeed high levels of unassembled viral proteins < 70 kDa, which can lead to a strong and persistent B cell receptor (BCR) activation, enhancing the Th2 immune response, releasing interleukin-10 (IL-10) and transforming growth factor-beta (TGF-β) that may lead to the immune paralysis, apoptosis, sepsis, and death. Finally, it is suggested that the polymerization of the viral antigens < 70 kDa into an antigenic polymer > 70 kDa could shift the immune response type from Th2 to Th1, developing the memory B cells and immunoglobulin G2 (IgG2) production, and avoiding the sepsis.
This review pretends to shed light on the immune processes occurring in the coronavirus disease 2019 (COVID-19) from a perspective based on the antigens size, lower or larger than 70 kDa. This cutoff size point explains the host type of immune response against the antigenic proteins of the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), which may lead to the development of the memory B cells or, conversely, the immune suppression, apoptosis, viral escape, and sepsis. Here, based on previous experimental work and the review of related literature, the following is proposed: antigens < 70 kDa can access the germinal center through the follicular conduits, where the activated B cells can present the processed antigen to specific naive CD4+ T cells that, in interaction with the major histocompatibility complex class II (MHC-II), trigger the immune response T helper type 2 (Th2). Conversely, antigens > 70 kDa cannot circulate through the narrow follicular conduits network and might be captured within the subcapsular sinus by the macrophages and dendritic follicular cells. Then, these cognate antigens are presented, via complement receptors, to the B cells that acquire and present them through the MHC-II to the specific naive CD4+ T cells, triggering the immune response Th1. The sustained infected cells lysis can overfeed high levels of unassembled viral proteins < 70 kDa, which can lead to a strong and persistent B cell receptor (BCR) activation, enhancing the Th2 immune response, releasing interleukin-10 (IL-10) and transforming growth factor-beta (TGF-β) that may lead to the immune paralysis, apoptosis, sepsis, and death. Finally, it is suggested that the polymerization of the viral antigens < 70 kDa into an antigenic polymer > 70 kDa could shift the immune response type from Th2 to Th1, developing the memory B cells and immunoglobulin G2 (IgG2) production, and avoiding the sepsis.
DOI: https://doi.org/10.37349/ei.2022.00061
This article belongs to the special issue The Sepsis induced Immune Conundrum
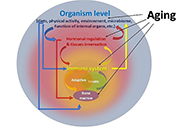
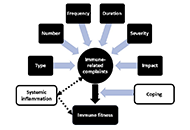
Interest in the mechanisms of aging of the immune system has not faded over the past 100 years, and it is caused by the immune-mediated development of age-related pathology, including autoimmune organ damage, reduced vaccination efficiency, atherosclerosis, the development of cardiovascular pathology, etc. In contrast to many other organs and systems, the immune system aging begins at an early age and has more pronounced changes that lead to the development of secondary pathology, which significantly affects life expectancy. But an effective strategy to restore immune function has not been developed yet. During this time, the mechanisms of age-related dysfunction of organs and cells of both the adaptive and innate immune systems were studied in detail—thymus involution, a decrease in the potential of hematopoietic stem cells, impaired differentiation and functions of immunocompetent cells, as well as the ways of their interaction. Numerous potential therapeutic targets have been identified and various approaches have been used to implement such therapeutic interventions. The review is devoted to replacement therapy using transplantation of hematopoietic stem cells (HSCs) and young lymphoid cells and tissues, cellular and systemic factor exchange in heterochronic parabiosis, and some other widely used life extension approaches. It has been proven that cell therapy using young cells to rejuvenate the old immune system, unfortunately, often turns out to be ineffective because it does not eliminate the root cause of age-related changes. The phenomenon of inflamm-aging that develops with age can significantly affect both the aging of the organism in general and the functioning of immunocompetent cells in particular. Therefore, the most promising direction in the restoration of immune functions during aging is systemic approaches that have a complex effect on the organism as a whole and can slow down the aging process.
Interest in the mechanisms of aging of the immune system has not faded over the past 100 years, and it is caused by the immune-mediated development of age-related pathology, including autoimmune organ damage, reduced vaccination efficiency, atherosclerosis, the development of cardiovascular pathology, etc. In contrast to many other organs and systems, the immune system aging begins at an early age and has more pronounced changes that lead to the development of secondary pathology, which significantly affects life expectancy. But an effective strategy to restore immune function has not been developed yet. During this time, the mechanisms of age-related dysfunction of organs and cells of both the adaptive and innate immune systems were studied in detail—thymus involution, a decrease in the potential of hematopoietic stem cells, impaired differentiation and functions of immunocompetent cells, as well as the ways of their interaction. Numerous potential therapeutic targets have been identified and various approaches have been used to implement such therapeutic interventions. The review is devoted to replacement therapy using transplantation of hematopoietic stem cells (HSCs) and young lymphoid cells and tissues, cellular and systemic factor exchange in heterochronic parabiosis, and some other widely used life extension approaches. It has been proven that cell therapy using young cells to rejuvenate the old immune system, unfortunately, often turns out to be ineffective because it does not eliminate the root cause of age-related changes. The phenomenon of inflamm-aging that develops with age can significantly affect both the aging of the organism in general and the functioning of immunocompetent cells in particular. Therefore, the most promising direction in the restoration of immune functions during aging is systemic approaches that have a complex effect on the organism as a whole and can slow down the aging process.
DOI: https://doi.org/10.37349/ei.2023.00105
This article belongs to the special issue Immunosenescence: Mechanisms and Its Impact
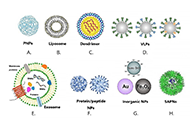
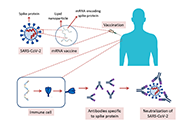
Vaccination has made an enormous contribution to global health. Treatment resistance for infectious diseases is growing quickly, and chemotherapeutic toxicity in cancer means that vaccines must be made right away to save humanity. But subunit vaccinations alone don’t give enough strong and long-lasting protection against infections that can kill. Nanoparticle (NP)-based delivery vehicles, such as dendrimers, liposomes, micelles, virosomes, nanogels, and microemulsions, offer interesting ways to get around the problems with traditional vaccine adjuvants. The nanovaccines (50–250 nm in size) are most efficient in terms of tissue targeting, staying in the bloodstream for a long time. Nanovaccines can improve antigen presentation, targeted delivery, stimulation of the body’s innate immune system, and a strong T-cell response without putting people at risk. This can help fight infectious diseases and cancers. Also, nanovaccines can be very helpful for making cancer treatments that use immunotherapy. So, this review highlights the various types of NPs used in the techniques that have worked in the new paradigm in viral vaccinology for infectious diseases. It gives a full rundown of the current NP-based vaccines, their potential as adjuvants, and the ways they can be delivered to cells. In the future, the best nanovaccines will try to be more logically designed, have more antigens in them, be fully functionalized, and be given to the right people.
Vaccination has made an enormous contribution to global health. Treatment resistance for infectious diseases is growing quickly, and chemotherapeutic toxicity in cancer means that vaccines must be made right away to save humanity. But subunit vaccinations alone don’t give enough strong and long-lasting protection against infections that can kill. Nanoparticle (NP)-based delivery vehicles, such as dendrimers, liposomes, micelles, virosomes, nanogels, and microemulsions, offer interesting ways to get around the problems with traditional vaccine adjuvants. The nanovaccines (50–250 nm in size) are most efficient in terms of tissue targeting, staying in the bloodstream for a long time. Nanovaccines can improve antigen presentation, targeted delivery, stimulation of the body’s innate immune system, and a strong T-cell response without putting people at risk. This can help fight infectious diseases and cancers. Also, nanovaccines can be very helpful for making cancer treatments that use immunotherapy. So, this review highlights the various types of NPs used in the techniques that have worked in the new paradigm in viral vaccinology for infectious diseases. It gives a full rundown of the current NP-based vaccines, their potential as adjuvants, and the ways they can be delivered to cells. In the future, the best nanovaccines will try to be more logically designed, have more antigens in them, be fully functionalized, and be given to the right people.
DOI: https://doi.org/10.37349/ei.2023.00107
This article belongs to the special issue Old and New Paradigms in Viral Vaccinology
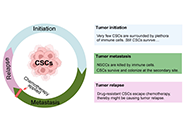
Cancer stem cells (CSCs) are a small subpopulation of cells that drive the formation and progression of tumors. However, during tumor initiation, how CSCs communicate with neighbouring immune cells to overcome the powerful immune surveillance barrier in order to form, spread, and maintain the tumor, remains poorly understood. It is, therefore, absolutely necessary to understand how a small number of tumor-initiating cells (TICs) survive immune attack during (a) the “elimination phase” of “tumor immune-editing”, (b) the establishment of regional or distant tumor after metastasis, and (c) recurrence after therapy. Mounting evidence suggests that CSCs suppress the immune system through a variety of distinct mechanisms that ensure the survival of not only CSCs but also non-stem cancer cells (NSCCs), which eventually form the tumor mass. In this review article, the mechanisms via which CSCs change the immune landscape of the tissue of origin, which contains macrophages, dendritic cells (DCs), myeloid-derived suppressor cells (MDSCs), natural killer (NK) cells, and tumor-infiltrating lymphocytes, in favour of tumorigenesis were discussed. The failure of cancer immunotherapy might also be explained by such interaction between CSCs and immune cells. This review will shed light on the critical role of CSCs in tumor immune evasion and emphasize the importance of CSC-targeted immunotherapy as a cutting-edge technique for battling cancer by restricting communication between immune cells and CSCs.
Cancer stem cells (CSCs) are a small subpopulation of cells that drive the formation and progression of tumors. However, during tumor initiation, how CSCs communicate with neighbouring immune cells to overcome the powerful immune surveillance barrier in order to form, spread, and maintain the tumor, remains poorly understood. It is, therefore, absolutely necessary to understand how a small number of tumor-initiating cells (TICs) survive immune attack during (a) the “elimination phase” of “tumor immune-editing”, (b) the establishment of regional or distant tumor after metastasis, and (c) recurrence after therapy. Mounting evidence suggests that CSCs suppress the immune system through a variety of distinct mechanisms that ensure the survival of not only CSCs but also non-stem cancer cells (NSCCs), which eventually form the tumor mass. In this review article, the mechanisms via which CSCs change the immune landscape of the tissue of origin, which contains macrophages, dendritic cells (DCs), myeloid-derived suppressor cells (MDSCs), natural killer (NK) cells, and tumor-infiltrating lymphocytes, in favour of tumorigenesis were discussed. The failure of cancer immunotherapy might also be explained by such interaction between CSCs and immune cells. This review will shed light on the critical role of CSCs in tumor immune evasion and emphasize the importance of CSC-targeted immunotherapy as a cutting-edge technique for battling cancer by restricting communication between immune cells and CSCs.
DOI: https://doi.org/10.37349/ei.2023.00108
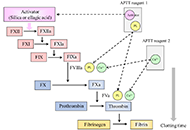
Antiphospholipid syndrome (APS) is defined as an autoimmune and prothrombotic disorder in patients with the persistent presence of antiphospholipid antibodies (aPLs). In the classification criteria, aPL expresses lupus anticoagulant (LA) activity, which is detected by prolongation of coagulation assays. The LA detection algorithm is a sequential flow including screening tests, mixing tests, and confirmatory tests to differentiate between LA-positive and other anticoagulant abnormalities. Two types of assays are used, like dilute Russell’s viper venom time (dRVVT) and activated partial thromboplastin time (APTT) because no single test is sensitive to all LAs. The anticoagulant drugs prescribed for the prevention and treatment of thrombosis disorders can interfere with the assays, and it is important to know the effects of these drugs in the assays. Especially, new generation anticoagulant drugs, called direct oral anticoagulants (DOACs), affect the results. In this review, the following points are discussed: i) LA detection flow and data interpretation, ii) the principles of coagulation assays proposed and their characteristics, and iii) the effects of anticoagulant drugs in LA detection.
Antiphospholipid syndrome (APS) is defined as an autoimmune and prothrombotic disorder in patients with the persistent presence of antiphospholipid antibodies (aPLs). In the classification criteria, aPL expresses lupus anticoagulant (LA) activity, which is detected by prolongation of coagulation assays. The LA detection algorithm is a sequential flow including screening tests, mixing tests, and confirmatory tests to differentiate between LA-positive and other anticoagulant abnormalities. Two types of assays are used, like dilute Russell’s viper venom time (dRVVT) and activated partial thromboplastin time (APTT) because no single test is sensitive to all LAs. The anticoagulant drugs prescribed for the prevention and treatment of thrombosis disorders can interfere with the assays, and it is important to know the effects of these drugs in the assays. Especially, new generation anticoagulant drugs, called direct oral anticoagulants (DOACs), affect the results. In this review, the following points are discussed: i) LA detection flow and data interpretation, ii) the principles of coagulation assays proposed and their characteristics, and iii) the effects of anticoagulant drugs in LA detection.
DOI: https://doi.org/10.37349/ei.2023.00110
This article belongs to the special issue Autoantibodies Associated to Thrombosis and Hemostasis
Vaccines are prophylactic medical products effectively used against infectious diseases. Although a high amount of vaccine studies are conducted at the preclinical stage, the number of approved vaccines is less than 10%. Development of vaccines from the research stage to the approval of administrative institutions takes about 5 years to 10 years conventionally. However, this period of time for vaccine development is not convenient during public health emergencies because an effective vaccine is required in a short time to restrict the speed of high mortality and morbidity. The pandemic of coronavirus disease 2019 (COVID-19), caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), had its catastrophic effects worldwide quickly. Therefore, an atypical process was followed for the development of COVID-19 vaccines. Great effort was spent in terms of cooperation among the governmental institutions, academia, and medical companies as well as a high amount of budget was allocated to develop effective vaccines against COVID-19. As of March 2023, the numbers of COVID-19 vaccines in clinical and preclinical development were 183 and 199, respectively. An emergency use authorization (EUA) process was applied to accelerate the approval of the vaccines. Consequently, vaccinations could be started in less than a year, which decelerated the speed of the pandemic. Although EUA caused hesitancy among some people questioning the safety and efficacy of the vaccines, the vast majority of the population was vaccinated. Currently, more than 5.5 billion people (about 70% of the world population) have received 13.38 billion doses of 11 different COVID-19 vaccines, and 73% of the doses were Comirnaty manufactured by Pfizer/BioNTech.
Vaccines are prophylactic medical products effectively used against infectious diseases. Although a high amount of vaccine studies are conducted at the preclinical stage, the number of approved vaccines is less than 10%. Development of vaccines from the research stage to the approval of administrative institutions takes about 5 years to 10 years conventionally. However, this period of time for vaccine development is not convenient during public health emergencies because an effective vaccine is required in a short time to restrict the speed of high mortality and morbidity. The pandemic of coronavirus disease 2019 (COVID-19), caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), had its catastrophic effects worldwide quickly. Therefore, an atypical process was followed for the development of COVID-19 vaccines. Great effort was spent in terms of cooperation among the governmental institutions, academia, and medical companies as well as a high amount of budget was allocated to develop effective vaccines against COVID-19. As of March 2023, the numbers of COVID-19 vaccines in clinical and preclinical development were 183 and 199, respectively. An emergency use authorization (EUA) process was applied to accelerate the approval of the vaccines. Consequently, vaccinations could be started in less than a year, which decelerated the speed of the pandemic. Although EUA caused hesitancy among some people questioning the safety and efficacy of the vaccines, the vast majority of the population was vaccinated. Currently, more than 5.5 billion people (about 70% of the world population) have received 13.38 billion doses of 11 different COVID-19 vaccines, and 73% of the doses were Comirnaty manufactured by Pfizer/BioNTech.
DOI: https://doi.org/10.37349/ei.2023.00111
This article belongs to the special issue Old and New Paradigms in Viral Vaccinology
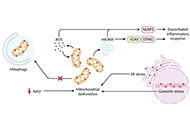
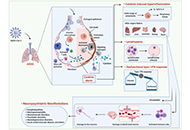
Immunosenescence encompasses multiple age-related adaptations that result in increased susceptibility to infections, chronic inflammatory disorders, and higher mortality risk. Macrophages are key innate cells implicated in inflammatory responses and tissue homeostasis, functions progressively compromised by aging. This process coincides with declining mitochondrial physiology, whose integrity is required to sustain and orchestrate immune responses. Indeed, multiple insults observed in aged macrophages have been implied as drivers of mitochondrial dysfunction, but how this translates into impaired immune function remains sparsely explored. This review provides a perspective on recent studies elucidating the underlying mechanisms linking dysregulated mitochondria homeostasis to immune function in aged macrophages. Genomic stress alongside defective mitochondrial turnover accounted for the progressive accumulation of damaged mitochondria in aged macrophages, thus resulting in a higher susceptibility to excessive mitochondrial DNA (mtDNA) leakage and reactive oxygen species (ROS) production. Increased levels of these mitochondrial products following infection were demonstrated to contribute to exacerbated inflammatory responses mediated by overstimulation of NOD-like receptor family pyrin domain containing 3 (NLRP3) inflammasome and cyclic GMP-ATP synthase (cGAS)-stimulator of interferon genes (STING) pathways. While these mechanisms are not fully elucidated, the present evidence provides a promising area to be explored and a renewed perspective of potential therapeutic targets for immunological dysfunction.
Immunosenescence encompasses multiple age-related adaptations that result in increased susceptibility to infections, chronic inflammatory disorders, and higher mortality risk. Macrophages are key innate cells implicated in inflammatory responses and tissue homeostasis, functions progressively compromised by aging. This process coincides with declining mitochondrial physiology, whose integrity is required to sustain and orchestrate immune responses. Indeed, multiple insults observed in aged macrophages have been implied as drivers of mitochondrial dysfunction, but how this translates into impaired immune function remains sparsely explored. This review provides a perspective on recent studies elucidating the underlying mechanisms linking dysregulated mitochondria homeostasis to immune function in aged macrophages. Genomic stress alongside defective mitochondrial turnover accounted for the progressive accumulation of damaged mitochondria in aged macrophages, thus resulting in a higher susceptibility to excessive mitochondrial DNA (mtDNA) leakage and reactive oxygen species (ROS) production. Increased levels of these mitochondrial products following infection were demonstrated to contribute to exacerbated inflammatory responses mediated by overstimulation of NOD-like receptor family pyrin domain containing 3 (NLRP3) inflammasome and cyclic GMP-ATP synthase (cGAS)-stimulator of interferon genes (STING) pathways. While these mechanisms are not fully elucidated, the present evidence provides a promising area to be explored and a renewed perspective of potential therapeutic targets for immunological dysfunction.
DOI: https://doi.org/10.37349/ei.2023.00112
This article belongs to the special issue Immunosenescence: Mechanisms and Its Impact
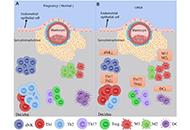
The etiology of recurrent spontaneous abortion (RSA) is extremely complex, as there are 40–50% of patients with unexplained miscarriages, known as unexplained RSA (URSA). URSA affects approximately 1–2% of females of childbearing age and has a massive impact on the physical and mental conditions of both patients and their families. The pathogenesis of the disease remains unclear, making its treatment complicated. In recent years, considerable progress has been made in the exploration of the URSA immune balance mechanism and it has been universally acknowledged that a balanced immune response (as abnormal immunity) may be the root cause of poor pregnancy outcomes. This review discussed and summarized the effects of immune cells and blocking antibodies (BAs) on URSA based on the current state of knowledge in this area. Additionally, molecular genetics also plays an essential role in the incidence rate of URSA since the role of genetic polymorphism in the pathogenesis of URSA has been thoroughly studied. Nonetheless, the outcomes of these studies are inconsistent, particularly across populations. This paper reviewed previous studies on URSA and maternal genetic polymorphism, focusing on and synthesizing the most important findings to date, and providing diagnostic recommendation for URSA patients with clinical symptoms.
The etiology of recurrent spontaneous abortion (RSA) is extremely complex, as there are 40–50% of patients with unexplained miscarriages, known as unexplained RSA (URSA). URSA affects approximately 1–2% of females of childbearing age and has a massive impact on the physical and mental conditions of both patients and their families. The pathogenesis of the disease remains unclear, making its treatment complicated. In recent years, considerable progress has been made in the exploration of the URSA immune balance mechanism and it has been universally acknowledged that a balanced immune response (as abnormal immunity) may be the root cause of poor pregnancy outcomes. This review discussed and summarized the effects of immune cells and blocking antibodies (BAs) on URSA based on the current state of knowledge in this area. Additionally, molecular genetics also plays an essential role in the incidence rate of URSA since the role of genetic polymorphism in the pathogenesis of URSA has been thoroughly studied. Nonetheless, the outcomes of these studies are inconsistent, particularly across populations. This paper reviewed previous studies on URSA and maternal genetic polymorphism, focusing on and synthesizing the most important findings to date, and providing diagnostic recommendation for URSA patients with clinical symptoms.
DOI: https://doi.org/10.37349/ei.2023.00113
Aim:
The study aims to evaluate the incidence of recurrent thromboses in patients with primary antiphospholipid syndrome (PAPS) and its association with the presence of different antiphospholipid antibodies (aPLs) and known thrombogenic risk factors.
Methods:
This retrospective study included 52 patients. The median age of the patients was 38.5 years [31.5; 43.5], and the duration of the disease was 9.0 years [3.1; 13.0]. aPLs, including IgG/IgM/IgA antibodies to cardiolipin (aCLs), IgG/IgM/IgA anti-beta2-glycoprotein I (anti-β2-GPI), IgG anti-domain I-β2-GPI (anti-β2-GPIDI) antibodies, IgG/IgM antibodies to the phosphatidylserine/prothrombin complex (aPS/PT), and other thrombosis risk factors were included for analysis.
Results:
Recurrent thrombosis was reported in 34 (65%) out of 52 patients and 18 (35%) did not have it. The main reason for the recurrence of thrombosis was the lack of anticoagulant therapy: in 18 (52.9%) out of 34 patients with recurrent thrombosis. Three patients were taking warfarin at the time of thrombosis recurrence, but target international normalized ratio (INR) levels were not achieved. Other patients with recurrent thrombosis were taking direct oral anticoagulants (DOACs). The risk of recurrent thrombotic events with positive IgG aCL was 10.33 (P = 0.002) and 21 (P = 0.007) times higher were examined in enzyme-linked immunoassay (ELISA) and chemiluminescent assay (CLA), respectively. The risk of thrombosis was 4.58 times higher in patients who were IgA aCL-positive (P = 0.01). Compared with other antibodies, with positive IgG values of anti-β2-GPI and IgG aPS/PT by ELISA, a lower probability of thrombosis recurrence was observed: 7.56 and 7.25, respectively. A high risk of recurrent thrombosis [odds ratio (OR) = 32.0] was observed in IgG anti-β2-GPI (CLA). The combination of IgG aCL with IgG anti-β2-GPI and with IgG anti-β2-GPIDI is more informative with respect to the risks of thrombosis recurrence compared to double positivity for aCL with anti-β2-GPI (OR = 20.71 vs. OR = 10.18). Triple positivity for IgG aCL with IgG anti-β2-GPI and with IgG aPS/PT also shows better results compared to positivity for aCL with anti-β2-GPI (OR = 6.06 vs. OR = 5.79). Among other risk factors, arterial hypertension (AH) and obesity were significant in relation to the recurrence of thrombosis. AH occurred in 22 (42%) of 52 patients with PAPS. AH was associated with recurrent thrombosis in PAPS patients: 18 (53%) out of 34 with recurrent thrombosis had AH versus 4 out of 18 without recurrent thrombosis (P = 0.003).
Conclusions:
Recurrent thrombosis in antiphospholipid syndrome (APS) is largely associated with IgG aCL, IgG anti-β2-GPI, IgG anti-β2-GPIDI, IgG aPS/PT, and IgA aCL positivity. AH was a significant risk factor for recurrent thrombosis.
Aim:
The study aims to evaluate the incidence of recurrent thromboses in patients with primary antiphospholipid syndrome (PAPS) and its association with the presence of different antiphospholipid antibodies (aPLs) and known thrombogenic risk factors.
Methods:
This retrospective study included 52 patients. The median age of the patients was 38.5 years [31.5; 43.5], and the duration of the disease was 9.0 years [3.1; 13.0]. aPLs, including IgG/IgM/IgA antibodies to cardiolipin (aCLs), IgG/IgM/IgA anti-beta2-glycoprotein I (anti-β2-GPI), IgG anti-domain I-β2-GPI (anti-β2-GPIDI) antibodies, IgG/IgM antibodies to the phosphatidylserine/prothrombin complex (aPS/PT), and other thrombosis risk factors were included for analysis.
Results:
Recurrent thrombosis was reported in 34 (65%) out of 52 patients and 18 (35%) did not have it. The main reason for the recurrence of thrombosis was the lack of anticoagulant therapy: in 18 (52.9%) out of 34 patients with recurrent thrombosis. Three patients were taking warfarin at the time of thrombosis recurrence, but target international normalized ratio (INR) levels were not achieved. Other patients with recurrent thrombosis were taking direct oral anticoagulants (DOACs). The risk of recurrent thrombotic events with positive IgG aCL was 10.33 (P = 0.002) and 21 (P = 0.007) times higher were examined in enzyme-linked immunoassay (ELISA) and chemiluminescent assay (CLA), respectively. The risk of thrombosis was 4.58 times higher in patients who were IgA aCL-positive (P = 0.01). Compared with other antibodies, with positive IgG values of anti-β2-GPI and IgG aPS/PT by ELISA, a lower probability of thrombosis recurrence was observed: 7.56 and 7.25, respectively. A high risk of recurrent thrombosis [odds ratio (OR) = 32.0] was observed in IgG anti-β2-GPI (CLA). The combination of IgG aCL with IgG anti-β2-GPI and with IgG anti-β2-GPIDI is more informative with respect to the risks of thrombosis recurrence compared to double positivity for aCL with anti-β2-GPI (OR = 20.71 vs. OR = 10.18). Triple positivity for IgG aCL with IgG anti-β2-GPI and with IgG aPS/PT also shows better results compared to positivity for aCL with anti-β2-GPI (OR = 6.06 vs. OR = 5.79). Among other risk factors, arterial hypertension (AH) and obesity were significant in relation to the recurrence of thrombosis. AH occurred in 22 (42%) of 52 patients with PAPS. AH was associated with recurrent thrombosis in PAPS patients: 18 (53%) out of 34 with recurrent thrombosis had AH versus 4 out of 18 without recurrent thrombosis (P = 0.003).
Conclusions:
Recurrent thrombosis in antiphospholipid syndrome (APS) is largely associated with IgG aCL, IgG anti-β2-GPI, IgG anti-β2-GPIDI, IgG aPS/PT, and IgA aCL positivity. AH was a significant risk factor for recurrent thrombosis.
DOI: https://doi.org/10.37349/ei.2023.00114
This article belongs to the special issue Autoantibodies Associated to Thrombosis and Hemostasis
One of the greatest challenges in the study of coronavirus disease 2019 (COVID-19) has been to establish the determining factors in the severity of the disease. Through extensive research efforts, a crucial factor responsible for disease control or exacerbation in COVID-19 has been identified—the regulation of the immune response. The abnormal release of interleukin-1 (IL-1), IL-6, and tumor necrosis factor-alpha (TNF-α) has been extensively studied in the context of the altered immune response observed in severe cases of COVID-19. However, recent attention has turned towards the excessive release of IL-17 and the increased presence of T helper 17 (Th17) cells, the main secretory cells of this cytokine. These factors have garnered interest due to their potential involvement in the cytokine storm observed in severe cases of COVID-19. In this review, it will be delved into the intricate mechanisms by which IL-6 contributes to the differentiation of Th17 cells, resulting in an increase in the population of Th17 cells. Moreover, it will be explored the proportional relationship between the increase of these lymphocytes and the release of IL-17 and other chemokines, which all together play a key role in promoting the chemotaxis and activation of neutrophils. Ultimately, this cascade of events culminates in the generation of tissue damage by neutrophils. Additionally, therapeutic options targeting these lymphocytes and cytokines are explored, providing insights into potential avenues for intervention.
One of the greatest challenges in the study of coronavirus disease 2019 (COVID-19) has been to establish the determining factors in the severity of the disease. Through extensive research efforts, a crucial factor responsible for disease control or exacerbation in COVID-19 has been identified—the regulation of the immune response. The abnormal release of interleukin-1 (IL-1), IL-6, and tumor necrosis factor-alpha (TNF-α) has been extensively studied in the context of the altered immune response observed in severe cases of COVID-19. However, recent attention has turned towards the excessive release of IL-17 and the increased presence of T helper 17 (Th17) cells, the main secretory cells of this cytokine. These factors have garnered interest due to their potential involvement in the cytokine storm observed in severe cases of COVID-19. In this review, it will be delved into the intricate mechanisms by which IL-6 contributes to the differentiation of Th17 cells, resulting in an increase in the population of Th17 cells. Moreover, it will be explored the proportional relationship between the increase of these lymphocytes and the release of IL-17 and other chemokines, which all together play a key role in promoting the chemotaxis and activation of neutrophils. Ultimately, this cascade of events culminates in the generation of tissue damage by neutrophils. Additionally, therapeutic options targeting these lymphocytes and cytokines are explored, providing insights into potential avenues for intervention.
DOI: https://doi.org/10.37349/ei.2023.00115
DOI: https://doi.org/10.37349/ei.2023.00116
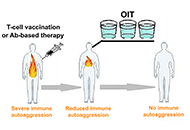
Immunotherapeutic treatment of autoimmune diseases should aim to inactivate autoaggressive memory T-cells and restore immune tolerance. It is envisaged that three approaches could be used to achieve this goal: stimulation of anti-idiotypic immune responses by vaccination with pathogenic T-cells; administration of suboptimal doses of antibodies (Abs) against two or more surface T-cell markers to provide selective Ab-mediated destruction of activated pathogenic memory T-cells; and induction of oral immune tolerance. The proposal entails the use of T-cell vaccination (TCV) or Ab-based therapy as an initial approach to reduce autoantigenic T-cell sensitization. Subsequently, the implementation of oral immunotherapy (OIT) is recommended to reinstate a consistent immune tolerance.
Immunotherapeutic treatment of autoimmune diseases should aim to inactivate autoaggressive memory T-cells and restore immune tolerance. It is envisaged that three approaches could be used to achieve this goal: stimulation of anti-idiotypic immune responses by vaccination with pathogenic T-cells; administration of suboptimal doses of antibodies (Abs) against two or more surface T-cell markers to provide selective Ab-mediated destruction of activated pathogenic memory T-cells; and induction of oral immune tolerance. The proposal entails the use of T-cell vaccination (TCV) or Ab-based therapy as an initial approach to reduce autoantigenic T-cell sensitization. Subsequently, the implementation of oral immunotherapy (OIT) is recommended to reinstate a consistent immune tolerance.
DOI: https://doi.org/10.37349/ei.2023.00117
Aim:
To describe the clinical characteristics and frequency of anti-factor H (FH) autoantibody-associated atypical hemolytic uremic syndrome (aHUS) in the first cohort of Argentine patients.
Methods:
The presence of anti-FH autoantibodies in 70 pediatric patients with suspected aHUS was investigated between 2013 and 2022. Clinical and laboratory parameters were collected and compared between patients who were positive and negative for anti-FH antibodies.
Results:
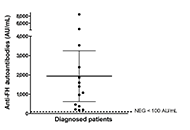
The 70 patients screened for anti-FH autoantibodies presented clinical features of non-immune microangiopathic hemolytic anemia, thrombocytopenia and renal injury. Positive titers were found in 14 children [mean: 1,938 arbitrary units per mL (AU/mL), range 179–8,500]. Due to missing clinical data, two patients who tested positive for anti-FH and 20 patients who tested negative for anti-FH were excluded from the data analysis. The laboratory features and clinical manifestations of anti-FH-positive aHUS cases (n = 12) were very similar to those of subjects with no autoantibodies detected (n = 36). Treatment administration was heterogeneous among the 12 patients analyzed. Dialysis was performed in six patients in total. Five children received plasmapheresis, while three patients were treated with plasma exchange followed by administration of eculizumab. Two patients received eculizumab only and one showed significant improvement solely through supportive care. Eight patients in total received immunosuppressive therapy. Follow-up of three patients showed a significant decrease of anti-FH autoantibody titers in 2/3 after treatment and during clinical remission.
Conclusions:
The cohort of 70 pediatric patients in this study demonstrated that the frequency of anti-FH autoantibody-associated aHUS in Argentina is 20%. The implementation of anti-FH testing in the country can potentially contribute to improved treatment and follow-up for patients with autoimmune aHUS.
Aim:
To describe the clinical characteristics and frequency of anti-factor H (FH) autoantibody-associated atypical hemolytic uremic syndrome (aHUS) in the first cohort of Argentine patients.
Methods:
The presence of anti-FH autoantibodies in 70 pediatric patients with suspected aHUS was investigated between 2013 and 2022. Clinical and laboratory parameters were collected and compared between patients who were positive and negative for anti-FH antibodies.
Results:
The 70 patients screened for anti-FH autoantibodies presented clinical features of non-immune microangiopathic hemolytic anemia, thrombocytopenia and renal injury. Positive titers were found in 14 children [mean: 1,938 arbitrary units per mL (AU/mL), range 179–8,500]. Due to missing clinical data, two patients who tested positive for anti-FH and 20 patients who tested negative for anti-FH were excluded from the data analysis. The laboratory features and clinical manifestations of anti-FH-positive aHUS cases (n = 12) were very similar to those of subjects with no autoantibodies detected (n = 36). Treatment administration was heterogeneous among the 12 patients analyzed. Dialysis was performed in six patients in total. Five children received plasmapheresis, while three patients were treated with plasma exchange followed by administration of eculizumab. Two patients received eculizumab only and one showed significant improvement solely through supportive care. Eight patients in total received immunosuppressive therapy. Follow-up of three patients showed a significant decrease of anti-FH autoantibody titers in 2/3 after treatment and during clinical remission.
Conclusions:
The cohort of 70 pediatric patients in this study demonstrated that the frequency of anti-FH autoantibody-associated aHUS in Argentina is 20%. The implementation of anti-FH testing in the country can potentially contribute to improved treatment and follow-up for patients with autoimmune aHUS.
DOI: https://doi.org/10.37349/ei.2023.00118
Since 2019, notable global viral outbreaks have occurred necessitating further research and healthcare system investigations. Following the coronavirus disease 2019 (COVID-19) pandemic, in 2022, whilst severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) strains evolved, monkeypox virus (MPXV) infections became more evident. MPXV is of the Orthopoxviridae genus, belonging to the family Poxviridae. Zoonotic transmission (animal-to-human transmission) may occur. The Orthopoxviridae genus includes other orthopoxviruses (OPXVs) present in animal host reservoirs that include cowpox viruses (CPXVs), vaccinia virus (VACV), and variola virus (VARV), with the latter being a causal agent of smallpox and excessive mortality. This review aims to present facts about MPXV-specific pathogenesis, epidemiology, and immunology alongside historical perspectives. MPXV was rarely reported outside Africa before April 2000. Early research since 1796 contributed towards the eradication of VARV leading to immunisation strategies. The World Health Organisation (WHO) announcement that VARV had been eradicated was confirmed in 1980. On the 23rd of July 2022, the WHO announced MPXV as a health emergency. Therefore, concern due to the propagation of MPXV causing monkeypox (mpox) disease requires clarity. Infected hosts display symptoms like extensive cellular-initiated rashes and lesions. Infection with MPXV makes it difficult to differentiate from other diseases or skin conditions. Antiviral therapeutic drugs were typically prescribed for smallpox and mpox disease; however, the molecular and immunological mechanisms with cellular changes remain of interest. Furthermore, no official authorized treatment exists for mpox disease. Some humans across the globe may be considered at risk. Historically, presenting symptoms of mpox resemble other viral diseases. Symptoms include rashes or lesions like Streptococcus, but also human herpes viruses (HHVs), including Varicella zoster virus (VZV).
Since 2019, notable global viral outbreaks have occurred necessitating further research and healthcare system investigations. Following the coronavirus disease 2019 (COVID-19) pandemic, in 2022, whilst severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) strains evolved, monkeypox virus (MPXV) infections became more evident. MPXV is of the Orthopoxviridae genus, belonging to the family Poxviridae. Zoonotic transmission (animal-to-human transmission) may occur. The Orthopoxviridae genus includes other orthopoxviruses (OPXVs) present in animal host reservoirs that include cowpox viruses (CPXVs), vaccinia virus (VACV), and variola virus (VARV), with the latter being a causal agent of smallpox and excessive mortality. This review aims to present facts about MPXV-specific pathogenesis, epidemiology, and immunology alongside historical perspectives. MPXV was rarely reported outside Africa before April 2000. Early research since 1796 contributed towards the eradication of VARV leading to immunisation strategies. The World Health Organisation (WHO) announcement that VARV had been eradicated was confirmed in 1980. On the 23rd of July 2022, the WHO announced MPXV as a health emergency. Therefore, concern due to the propagation of MPXV causing monkeypox (mpox) disease requires clarity. Infected hosts display symptoms like extensive cellular-initiated rashes and lesions. Infection with MPXV makes it difficult to differentiate from other diseases or skin conditions. Antiviral therapeutic drugs were typically prescribed for smallpox and mpox disease; however, the molecular and immunological mechanisms with cellular changes remain of interest. Furthermore, no official authorized treatment exists for mpox disease. Some humans across the globe may be considered at risk. Historically, presenting symptoms of mpox resemble other viral diseases. Symptoms include rashes or lesions like Streptococcus, but also human herpes viruses (HHVs), including Varicella zoster virus (VZV).
DOI: https://doi.org/10.37349/ei.2023.00119
This article belongs to the special issue Old and New Paradigms in Viral Vaccinology
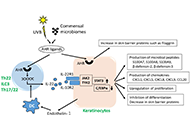
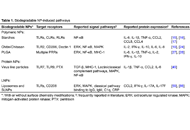
Interleukin (IL)-22 is produced from immune cells such as T helper (Th)22 cells, Th17/22 cells, and group 3 innate lymphoid cells. IL-22 signals via the IL-22 receptor 1 (IL-22R1) and the IL-10 receptor 2 (IL-10R2). As the IL-22R1/IL-10R2 heterodimer is preferentially expressed on border tissue between the host and the environment, IL-22 is believed to be involved in border defense. Epidermal keratinocytes are the first-line skin barrier and express IL-22R1/IL-10R2. IL-22 increases keratinocyte proliferation but inhibits differentiation. Aryl hydrocarbon receptor (AHR) is a chemical sensor and an essential transcription factor for IL-22 production. In addition, AHR also upregulates the production of barrier-related proteins such as filaggrin in keratinocytes, suggesting a pivotal role for the AHR-IL-22 axis in regulating the physiological skin barrier. Although IL-22 signatures are elevated in atopic dermatitis and psoriasis, their pathogenic and/or protective implications are not fully understood.
Interleukin (IL)-22 is produced from immune cells such as T helper (Th)22 cells, Th17/22 cells, and group 3 innate lymphoid cells. IL-22 signals via the IL-22 receptor 1 (IL-22R1) and the IL-10 receptor 2 (IL-10R2). As the IL-22R1/IL-10R2 heterodimer is preferentially expressed on border tissue between the host and the environment, IL-22 is believed to be involved in border defense. Epidermal keratinocytes are the first-line skin barrier and express IL-22R1/IL-10R2. IL-22 increases keratinocyte proliferation but inhibits differentiation. Aryl hydrocarbon receptor (AHR) is a chemical sensor and an essential transcription factor for IL-22 production. In addition, AHR also upregulates the production of barrier-related proteins such as filaggrin in keratinocytes, suggesting a pivotal role for the AHR-IL-22 axis in regulating the physiological skin barrier. Although IL-22 signatures are elevated in atopic dermatitis and psoriasis, their pathogenic and/or protective implications are not fully understood.
DOI: https://doi.org/10.37349/ei.2021.00005
This article belongs to the special issue Cross Talk Among Skin Cells and Immune Cells
Immunity is continuously evolving by evolutionary mechanisms shaped by pathogenic stimuli of different kinds. Man-made nanomaterials (NMs) have been developed in the last decades and represent a novel challenge for our immune system, especially when applied to medical science. Toxicological studies of such nanoparticles (NPs) revealed that size, shape, and surface chemistry are key parameters to understand their noxious effects on cellular mechanisms. Less is known on the immune reactions to NMs since prolonged exposure data are not so detailed as the results for acute administration. The importance of immunity to biocompatible NPs is underlined by their increasing use as drug or gene delivery carriers in common pharmaceutical preparations and vaccines. In the latter case, the immunomodulatory properties of NMs allow their use also as efficient adjuvants to enhance the innate immune response. In the current manuscript, the authors discuss the main concepts in this fast-growing field by restricting our view to NMs with consolidated application in biomedicine.
Immunity is continuously evolving by evolutionary mechanisms shaped by pathogenic stimuli of different kinds. Man-made nanomaterials (NMs) have been developed in the last decades and represent a novel challenge for our immune system, especially when applied to medical science. Toxicological studies of such nanoparticles (NPs) revealed that size, shape, and surface chemistry are key parameters to understand their noxious effects on cellular mechanisms. Less is known on the immune reactions to NMs since prolonged exposure data are not so detailed as the results for acute administration. The importance of immunity to biocompatible NPs is underlined by their increasing use as drug or gene delivery carriers in common pharmaceutical preparations and vaccines. In the latter case, the immunomodulatory properties of NMs allow their use also as efficient adjuvants to enhance the innate immune response. In the current manuscript, the authors discuss the main concepts in this fast-growing field by restricting our view to NMs with consolidated application in biomedicine.
DOI: https://doi.org/10.37349/ei.2021.00006
As the severe acute respiratory syndrome coronavirus (SARS-CoV)-2 is a new virus, the current knowledge on the immunopathogenesis of this newly emerged SARS-CoV-2 is beginning to unravel with intensive ongoing global research efforts. Although a plethora of new studies have been published in a short space of time describing how the virus causes disease and incurs insults on the host immune system and the underlying immunopathogenic mechanisms remain to be elucidated. Thus, the discussion in this review is based on the most current knowledge on the immunopathogenesis of SARS-CoV-2 that has emerged in the past 12 months. The main objective is to shed light on the most current concepts in immunopathological aspects of the lung, bloodstream, and brain caused by the SARS-CoV-2, which has led to the current pandemic resulting in > 100 million infections and > 2 million deaths, and ongoing.
As the severe acute respiratory syndrome coronavirus (SARS-CoV)-2 is a new virus, the current knowledge on the immunopathogenesis of this newly emerged SARS-CoV-2 is beginning to unravel with intensive ongoing global research efforts. Although a plethora of new studies have been published in a short space of time describing how the virus causes disease and incurs insults on the host immune system and the underlying immunopathogenic mechanisms remain to be elucidated. Thus, the discussion in this review is based on the most current knowledge on the immunopathogenesis of SARS-CoV-2 that has emerged in the past 12 months. The main objective is to shed light on the most current concepts in immunopathological aspects of the lung, bloodstream, and brain caused by the SARS-CoV-2, which has led to the current pandemic resulting in > 100 million infections and > 2 million deaths, and ongoing.
DOI: https://doi.org/10.37349/ei.2021.00007
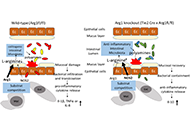
Arginase-1 (Arg1) and the inducible nitric oxide synthase 2 (NOS2) compete for the common substrate L-arginine, semi-essential amino acid, and central intestinal metabolite. Both enzymes exhibit various, sometimes opposing effects on immune responses, tissue regeneration, or microbial growth and replication. In sub-mucosal tissues of patients suffering from inflammatory bowel disease (IBD), similar as in experimental colitis, the expression and activity of both enzymes, Arg1 and NOS2 are more prominent than in respective controls. Accordingly, the metabolism of L-arginine is altered in IBD patients. Thus, L-arginine represents a promising medical target for clinical intervention in these devastating diseases. Previous studies primarily focused on the host side of L-arginine metabolism. Initial reports using Arg1 inhibitors generated conflicting results in murine colitis models. Subsequently, only the generation of conditional Arg1 knockout mice allowed reliable functional analyses of Arg1 and the L-arginine metabolism in the immune system. Utilizing cell-specific conditional Arg1 knockouts, we have recently reported that Arg1, surprisingly, hampered the resolution of experimental colitis due to the restriction of the intraluminal availability of L-arginine. Reduced levels of L-arginine restrained the compositional diversity of the intestinal microbiota and subsequently the mutual metabolism between the microbiota and the host. Thus, the intraluminal microbiota represents a potential therapeutic target for L-arginine metabolism aside from host-dependent L-arginine consumption.
Arginase-1 (Arg1) and the inducible nitric oxide synthase 2 (NOS2) compete for the common substrate L-arginine, semi-essential amino acid, and central intestinal metabolite. Both enzymes exhibit various, sometimes opposing effects on immune responses, tissue regeneration, or microbial growth and replication. In sub-mucosal tissues of patients suffering from inflammatory bowel disease (IBD), similar as in experimental colitis, the expression and activity of both enzymes, Arg1 and NOS2 are more prominent than in respective controls. Accordingly, the metabolism of L-arginine is altered in IBD patients. Thus, L-arginine represents a promising medical target for clinical intervention in these devastating diseases. Previous studies primarily focused on the host side of L-arginine metabolism. Initial reports using Arg1 inhibitors generated conflicting results in murine colitis models. Subsequently, only the generation of conditional Arg1 knockout mice allowed reliable functional analyses of Arg1 and the L-arginine metabolism in the immune system. Utilizing cell-specific conditional Arg1 knockouts, we have recently reported that Arg1, surprisingly, hampered the resolution of experimental colitis due to the restriction of the intraluminal availability of L-arginine. Reduced levels of L-arginine restrained the compositional diversity of the intestinal microbiota and subsequently the mutual metabolism between the microbiota and the host. Thus, the intraluminal microbiota represents a potential therapeutic target for L-arginine metabolism aside from host-dependent L-arginine consumption.
DOI: https://doi.org/10.37349/ei.2021.00008
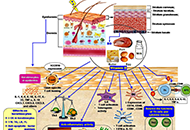
Skin is the largest organ of the body having multifunctional activities. It has a dynamic cellular network with unique immunologic properties to maintain defensive actions, photoprotection, immune response, inflammation, tolerogenic capacity, wound healing, etc. The immune cells of the skin exhibit distinct properties. They can synthesize active vitamin D [1,24(OH)2D3] and express vitamin D receptors. Any difficulties in the cutaneous immune system cause skin diseases (psoriasis, vitiligo, atopic dermatitis, skin carcinoma, and others). Vitamin D is an essential factor, exhibits immunomodulatory effects by regulating dendritic cells’ maturation, lymphocytes’ functions, and cytokine production. More specifically, vitamin D acts as an immune balancing agent, inhibits the exaggeration of immunostimulation. This vitamin suppresses T-helper 1 and T-helper 17 cell formation decreases inflammatory cytokines release and promotes the maturation of regulatory T cells and interleukin 10 secretion. The deficiency of this vitamin promotes the occurrence of immunoreactive disorders. Administration of vitamin D or its analogs is the therapeutic choice for the treatment of several skin diseases.
Skin is the largest organ of the body having multifunctional activities. It has a dynamic cellular network with unique immunologic properties to maintain defensive actions, photoprotection, immune response, inflammation, tolerogenic capacity, wound healing, etc. The immune cells of the skin exhibit distinct properties. They can synthesize active vitamin D [1,24(OH)2D3] and express vitamin D receptors. Any difficulties in the cutaneous immune system cause skin diseases (psoriasis, vitiligo, atopic dermatitis, skin carcinoma, and others). Vitamin D is an essential factor, exhibits immunomodulatory effects by regulating dendritic cells’ maturation, lymphocytes’ functions, and cytokine production. More specifically, vitamin D acts as an immune balancing agent, inhibits the exaggeration of immunostimulation. This vitamin suppresses T-helper 1 and T-helper 17 cell formation decreases inflammatory cytokines release and promotes the maturation of regulatory T cells and interleukin 10 secretion. The deficiency of this vitamin promotes the occurrence of immunoreactive disorders. Administration of vitamin D or its analogs is the therapeutic choice for the treatment of several skin diseases.
DOI: https://doi.org/10.37349/ei.2021.00009
This article belongs to the special issue Cross Talk Among Skin Cells and Immune Cells
