Review
Review

Affiliation:
Faculty of Medicine, Universiti Sultan Zainal Abidin, Kuala Terengganu 20400, Malaysia
ORCID: https://orcid.org/0009-0001-9190-6216
Affiliation:
Faculty of Medicine, Universiti Sultan Zainal Abidin, Kuala Terengganu 20400, Malaysia
ORCID: https://orcid.org/0000-0002-4194-7650
Affiliation:
Faculty of Medicine, Universiti Sultan Zainal Abidin, Kuala Terengganu 20400, Malaysia
Email: azuinsuliman@unisza.edu.my
ORCID: https://orcid.org/0000-0003-0296-3313
Explor Immunol. 2025;5:1003208 DOl: https://doi.org/10.37349/ei.2025.1003208
Received: February 10, 2025 Accepted: July 25, 2025 Published: August 11, 2025
Academic Editor: Manuela Neuman, University of Toronto, Canada
Rheumatoid arthritis (RA) is an inflammatory autoimmune disorder characterised by synovial joint destruction and systemic complications. Central to its pathogenesis is the formation and deposition of immune complexes (ICs), which result from antigen-antibody interactions involving autoantibodies such as rheumatoid factor (RF) and anti-citrullinated protein antibodies (ACPAs). These ICs infiltrate joint tissues, activate the complement system, and initiate a cascade of inflammatory responses. The ensuing recruitment of polymorphonuclear leukocytes and release of pro-inflammatory cytokines and chemokines contribute to sustained inflammation, tissue degradation, and joint deformity. RA is thus classified as a type III hypersensitivity disorder, wherein IC-mediated mechanisms perpetuate a self-amplifying inflammatory loop. This review explores the evolving understanding of IC-driven pathophysiology in RA, emphasising the three-stage progression of IC formation, deposition, and inflammatory activation. By elucidating the interplay between hypersensitivity reactions and immune-mediated mechanisms in RA, the review underscores potential therapeutic targets that may help disrupt this pathogenic cycle. Enhanced comprehension of IC dynamics not only deepens insight into RA progression but also opens avenues for more precise and effective interventions in autoimmune diseases.
Rheumatoid arthritis (RA) is a multisystem and chronic inflammatory autoimmune disease primarily affecting the joints. Affecting approximately 0.2% of the world population, the aetiology of RA is unknown. A meta-analysis done in 2020 revealed that the prevalence of RA increased to 0.46% of the world population between 1980 and 2019 [1]. The progression of RA can cause deformities and physical impairments if not treated appropriately [2]. RA also comprises the symptoms of inflammatory arthropathy, synovial hyperplasia, bone destruction, articular cartilage loss, and bone deformities. Immunologically, RA causes the production of autoantibodies such as rheumatoid factor (RF) and anti-citrullinated protein antibodies (ACPAs) [2, 3]. Besides physical and immunological presentation, extra-articular symptoms of RA are also presented by nodules, pericarditis, pulmonary fibrosis, peripheral neuropathy, and amyloidosis. RA’s extended effect indirectly affects the cardiovascular, pulmonary, neurological, and endocrine systems [2].
The mechanisms underlying the development of autoimmune diseases, caused by the immune system’s failure to protect an organism against pathogens while avoiding self-destruction, remain unclear [4]. Though the exact cause of RA is unknown, specific immune response pathways involving immune and non-immune cells secrete proinflammatory mediators, such as cytokines and enzymes, that disrupt the immune balance [5]. The most prevalent multiple autoantibodies in RA patients are RF, ACPAs, and antibodies against additional post-translationally modified proteins, such as anti-acetylated and anti-carbamylated protein (anti-CarP) antibodies [6]. The development of the immune response is associated with the interactions between established internal and external risk factors [3, 6]. Furthermore, the production of autoantibodies that target the joints of the entire body is one of the critical reasons for RA onset [7].
Immune complexes (ICs) are antigen-antibody aggregations that develop when the body’s immune system produces antibodies in response to antigenic determinants of host or foreign substances [8]. Usually, the cellular immune system removes excess ICs, protecting the tissues against immune-induced destruction [9]. Deposition of ICs in tissues due to insufficient clearance results in IC-mediated disease, known as type III hypersensitivity reaction [10]. Its responses involve IgG and IgM antibodies—the antigen-antibody complex deposits in the capillaries, surrounding tissues, and basement membrane [11]. Overproduction of immunoglobulin (IgM and IgG) against foreign or host antigens mediates the deposition of insoluble intermediate-sized ICs. These complexes in observed in type III hypersensitivity reactions. Subsequently, it may elicit activation of the classical complement, resulting in an overproduction of various inflammatory mediators, leading to progressive damage [10].
This review aims to synthesise recent advances in the understanding of IC-mediated mechanisms in RA, particularly within the framework of type III hypersensitivity reactions. To ensure relevance and rigor, we conducted a literature search using four major databases, which were PubMed, Web of Science, Scopus, and the Cochrane Library, focusing on publications from 2010 to 2025. Keyword combinations included “rheumatoid arthritis”, “immune complex”, “type III hypersensitivity”, and “autoantibodies”. Studies were selected based on their focus on the immunopathogenesis of RA, the role of ICs, and relevance to disease progression. By exploring the intersection of IC biology and hypersensitivity responses, this review offers insights into the evolving landscape of RA pathogenesis and highlights potential therapeutic targets aimed at disrupting the IC-driven inflammatory loop.
The immune system is essential for protecting the body against infections; however, in certain contexts, its responses can become exaggerated, misdirected, or dysregulated, leading to tissue injury rather than protection. These harmful immune reactions are collectively known as hypersensitivity reactions, defined as pathological immune responses to normally harmless antigens or to self-antigens, often resulting in persistent inflammation and tissue damage [12]. Hypersensitivity reactions arise from the same mechanisms that eliminate pathogens, including antibody production, T lymphocyte activation, and recruitment of effector cells. However, the immune stimuli in hypersensitivity, such as persistent environmental antigens, microbial components, or self-antigens, are often difficult to clear. The presence of intrinsic positive feedback loops within the immune system amplifies responses and contributes to the chronicity and severity of tissue damage once a hypersensitivity reaction is triggered [13].
Tissue injury in hypersensitivity response is caused by the exact mechanisms that regularly act to eradicate infectious pathogenic microorganisms with the assistance of antibodies, effector T lymphocytes, and various other effector cells. This is because the stimuli for these abnormal immune responses, such as self-antigen, persistent microbes, or environmental antigens, are challenging to eliminate. Moreover, the immune system has several intrinsic positive feedback loops, which commonly enhance protective immunity, making it difficult to control or terminate a hypersensitivity reaction once initiated [12].
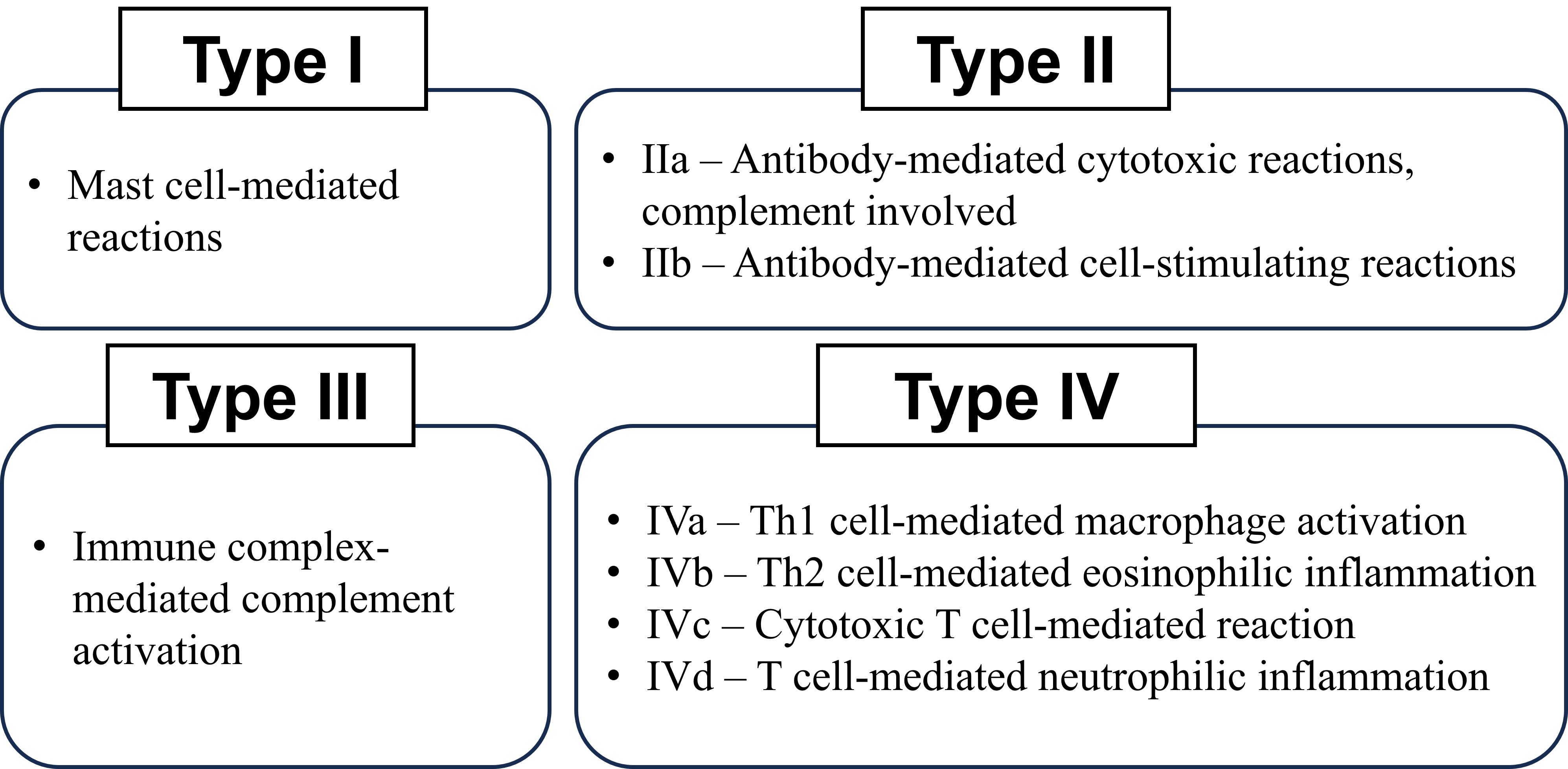
Since inflammation is a prominent component of the pathology of these diseases, they are grouped under the term immune-mediated inflammatory illness. Based on the principal immunological mechanism responsible for the condition, hypersensitivity reactions may be categorised into four types: the first three categories are variants on antibody-mediated, and the fourth is T-cell mediated [12]. The type of immune response and the effector mechanism responsible for cell and tissue injury are often used to classify hypersensitivity disease [14]. An article by Dispenza [15] proposed a more refined classification of hypersensitivity subtypes, incorporating effector mechanisms such as mast cell activation, complement involvement, cytotoxic T cell-mediated responses, and eosinophilic inflammation (Figure 1).
Over the past decade, multiple studies have linked different hypersensitivity mechanisms to the pathogenesis of autoimmune diseases, including RA. Research articles have speculated that the involvement of type II, type III [16], and even type IV hypersensitivity reactions [17] correlated with RA occurrences. However, this review article will focus on the involvement of type III hypersensitivity reactions as an inducer of RA incidence.
Type III hypersensitivity reaction features include the development of ICs triggered by the interaction of IgG or IgM with a soluble antigen [4, 13]. Type III hypersensitivity reactions are an effect of normal or abnormal immunisation exhibited in various autoimmune illnesses. An entry of a significant antigen concentration into the bloodstream mediates the generalised type III hypersensitivity reactions [4]. In contrast to the other hypersensitivity reactions, the type III reaction involves the production of ICs in the circulation before deposition in tissues [13].
In pathogenic circumstances, the innate immune system causes local and systemic inflammation, which destroys self-tissues. Conversely, in some cases, the immune system exaggerates, forcing the hosts to become hypersensitive to the excess production of antigen-antibody IC [4]. Circulated ICs tend to accumulate in filtration sites, such as in synovial joints, while the other ICs, generated at specific inoculation sites, enhance the stimulation of the classical complement pathway [13, 18]. The rapid development of symptoms and proliferation of ICs, combined with phagocytes and complement components, indicate the involvement of nonspecific or innate immune systems. As the latter initiates the type III hypersensitive mechanisms, it also leads to tissue damage by mediating the accumulation of inflammatory cells, lysosomal enzymes, and free radicals around the ICs. Inflammation within the walls of blood arteries, caused by complement activation and binding of leukocyte fragment crystallisable receptor (FcR) to antibodies in deposited complexes, is the principal cause of tissue harm in IC disorders [13, 19].
A hallmark feature of type III hypersensitivity is vascular inflammation (vasculitis), caused by FcR-mediated leukocyte activation and complement component binding to deposited ICs within blood vessel walls. This mechanism underpins a range of systemic IC-driven diseases [14], including RA, systemic lupus erythematosus (SLE), and serum sickness. The pathophysiological sequence is illustrated in Figure 2.
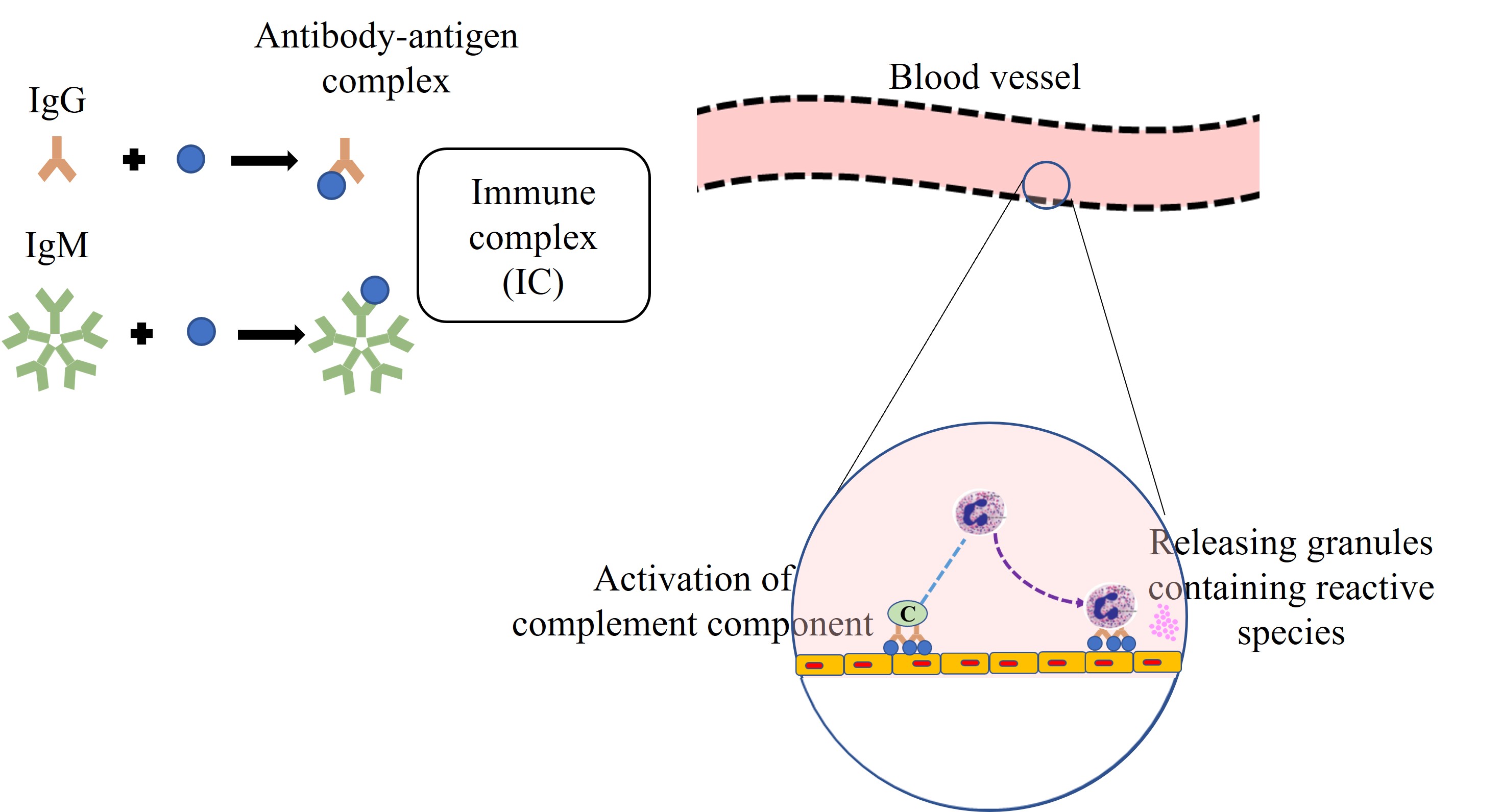
RA, an autoimmune disease, exhibits the clinical features of type III hypersensitivity reactions [13–15]. The reaction occurs due to the build-up of the ICs involving the autoantibodies recognising the soluble autoantigens, which are always present naturally and abundantly, besides the foreign antigens. The involvement of IgG is reported to relate to the incidence of RA [18]. Recent insights have confirmed that the dysregulated clearance of ICs in RA contributes to ongoing inflammation, suggesting that RA is not only autoimmune but also fundamentally IC-mediated [20–22].
Generally, mononuclear phagocytes clear off the ICs, which have been fixed by the complement cascade. The failure in the clearance of IC may result in IC-mediated disease [23]. The mononuclear phagocytes prevent IC deposition by enhancing their binding to erythrocytes, subsequently assisting in carrying the ICs to the spleen and liver for disposal by resident phagocytes [23, 24]. However, an insufficient amount of complement caused by genetic, antibody-mediated, or drug-induced factors may result in the accumulation of ICs and, thus, stimulate a chronic immune response. Besides, this condition can promote the formation of autoantibodies on the cell surface and intracellular antigens of nuclear or cytoplasmic origin. It can also activate systemic forms of autoimmune diseases such as RA [23].
ICs were discovered in RA 50 years ago, mostly in synovial fluid and the circulation of individuals with extra-articular disease [25]. The autoantibodies and ICs were fundamental to early theories in the pathogenesis of RA by sequentially activating the neutrophils and mast cells, allowing antibodies accessibility to the joints, where they would initiate inflammation by binding to a specific antigen [26]. RA is regarded as evidence of an innate immunity disorder, along with IC-mediated complement activation, adaptive immune responses against host-antigens composed primarily of post-translationally modified proteins, dysregulated cytokine networks, osteoclast activation, and activation of chondrocyte and resident stromal cells, which establish the semi-autonomous features that enhance the progression of disease [27]. The pathogenesis of RA, a systemic IC disease, can be categorised into three significant stages: formation of IC, deposition of IC, and IC-mediated inflammation and tissue damage [12, 13].
This review was performed following an extensive literature search utilising four principal scientific databases: PubMed, Web of Science, Scopus, and the Cochrane Library. The search focused on articles published from 2010 to 2025 to ensure an inclusion of recent and relevant findings. A combination of specific keywords was employed to extract essential literature, including: “immune complex”, “rheumatoid arthritis”, “hypersensitivity”, and “immunology of rheumatoid arthritis”.
A protein antigen causes an immunological response that produces antibodies within 4–10 days. The immunological reaction is then activated by the antibody interaction with the antigen, which forms ICs. Antibody production in the blood circulation is commonly triggered by endogenous or exogenous antigen exposure [12, 13]. Exogenous antigens are foreign proteins, whereas endogenous antigens are autoantigens that mimic foreign antigens directed at autoimmune diseases such as RA [12, 13, 28]. In both types of antigens, circulating ICs will be formed by binding antigens to antibodies, which later move out of the plasma and deposit in host tissues [13].
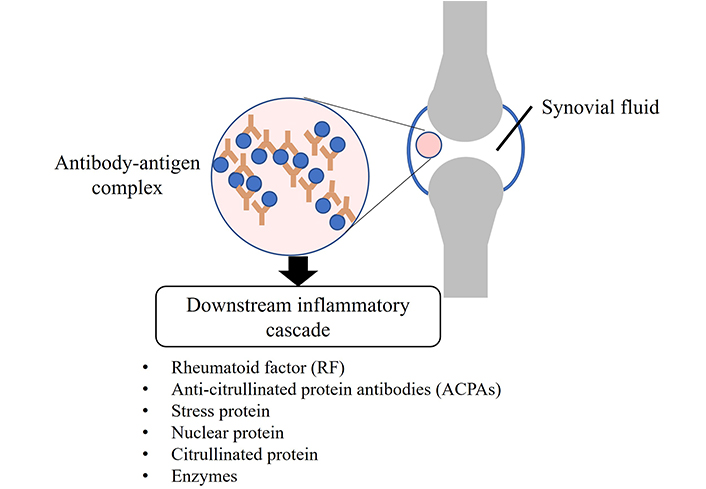
IC in joints can be formed by the autoantibodies observed in RA patients’ serum and synovial fluid [29]. The binding of autoantibodies to their specific target antigen forms the IC that triggers a downstream inflammatory cascade [5, 30]. RF and ACPAs are the autoantibodies identified and recognised as biomarkers for diagnosing RA [29, 31]. Autoantigens directed by several autoantibodies later can be discovered in RA, including a diverse range of components in cartilage, such as stress proteins, nuclear proteins, citrullinated proteins, and enzymes (illustrated in Figure 3). This accumulation of elements in RA demonstrates that the disease is characterised by accumulated autoreactivities in both B and T cells. These autoantigens and immunologically relevant epitopes vary in range during the disease course, and individuals’ self-antigens may differ [32, 33].
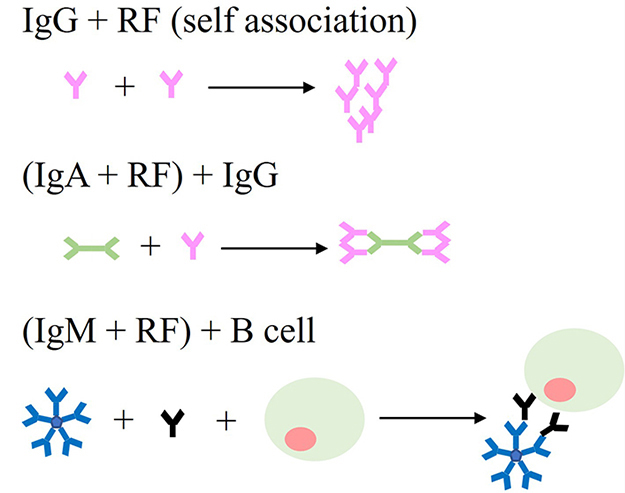
The initial autoantibodies that have been characterised in RA are RF, as a target to the Fc region of IgG, which is induced by and reactive towards IC [18, 34]. Three types of Ig are directly and indirectly involved in exhibiting RA: IgG [35], IgA [36], and IgM [37], summarised in Figure 4. The IgG RF antibodies are unique as they may form ICs by self-association without the presence of different antigenic molecules [36]. The self-association characterisation of IgG RFs has a significant implication for pathogenic disease progression in RA patients. Moreover, IgA RFs have also been identified in RA patients. By interacting with normal IgG, these polymeric IgA RFs contributed to the development of intermediate complexes [36]. Furthermore, the most common RF species in RA are IgM RFs, formed by ICs and polyclonal B-cell activators [34, 38]. Polyclonal low-affinity IgM RF formation is not disease-specific, but it is considered a normal physiological response that assists host defence by accelerating the ICs’ development and clearance. Though high-affinity RFs do not appear to induce RA, they are considered to contribute effectively to disease progression and chronicity by stimulating IC development. This is because RFs are produced by IC derived from disease-specific autoantibodies, which also facilitate the formation of IC and enhance the arthritogenicity of disease-specific autoantibodies, including ACPAs [34].
The development of IC between citrullinated proteins and ACPAs, advanced by complement fixation, is significant in RA synovium [39]. ACPAs are RA-specific and target epitopes focused on citrulline, a post-translationally modified arginine. The IC and citrullinated protein comprising different citrullinated antigens are more immunogenic and arthritogenic, while their existence in arthritic joints corresponds with the severity of the disease [34, 40]. Autoantibodies to type II collagen (CII) are also detected in RA and may develop IC in the joint as an essential factor in the early local onset of inflammation. The CII epitopes are found on the synovium and cartilage, while the autoantibodies to CII are released by synovial fluid cells and tissues in RA patients [41, 42] as illustrated in Figure 5. Besides, K/BxN mice, the most used autoimmune arthritis model, secrete antibodies against glucose-6-phosphate isomerase (GPI) that form ICs with GPI in the joints. These ICs also trigger arthritis in the same way as IC-mediated synovitis, considered to contribute to RA in humans [43, 44].
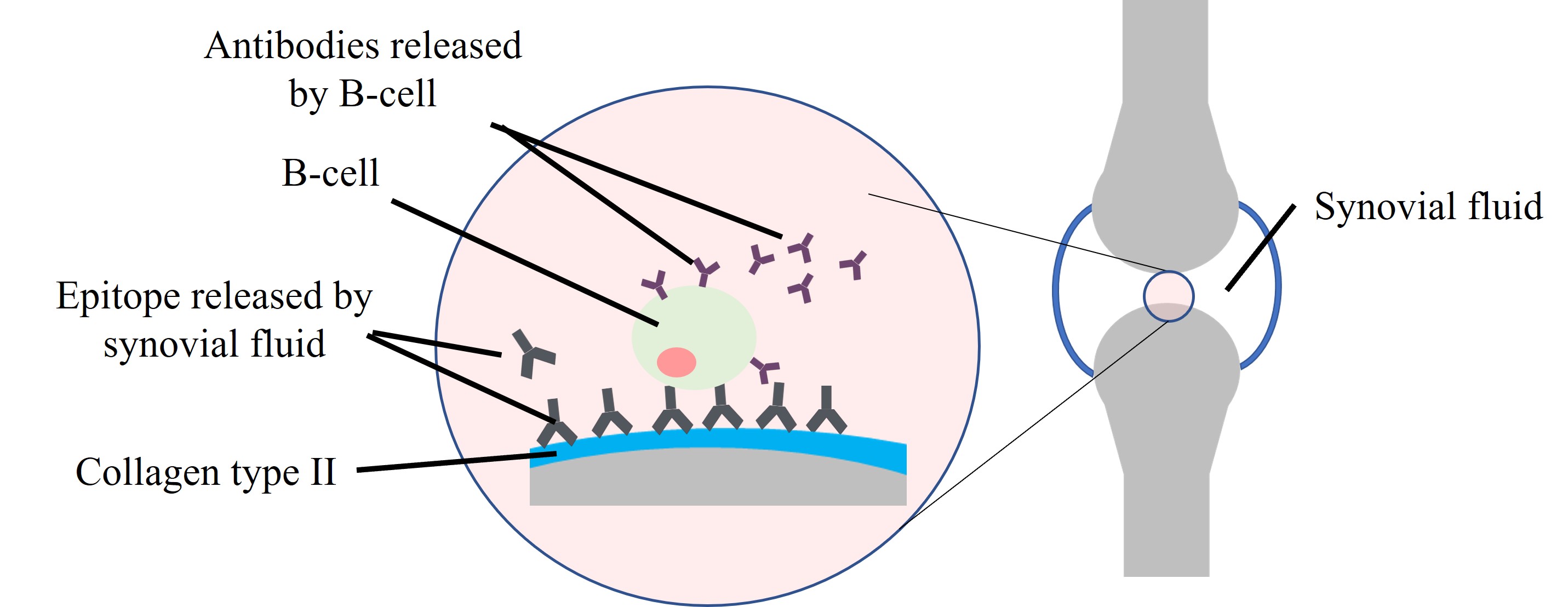
In chronic inflammatory arthritis, autoantibodies targeting antigens in the inflamed joint worsen inflammation by continuously producing ICs. Thus, persistent generation of ICs becomes the indicator of RA chronicity [35]. These excessively formed ICs potentially deposit in tissues, inducing inflammatory damage and the release of autoantigens and activating an autoimmune response [18].
The major histocompatibility complex (MHC), known as human leukocyte antigen (HLA) in humans, plays a crucial role in distinguishing self from foreign antigens [45]. While theories on immune responses to altered self-antigens or cross-reactivity are plausible, there is limited mechanistic and epidemiological evidence supporting them. Moreover, HLA alleles are linked to multiple immune-related diseases affecting different tissues, raising questions about the specificity of antigen presentation [18, 46].
HLA class II alleles are strongly associated with autoimmune diseases, particularly seropositive RA [47]. During T cell development, self-reactive T cells are eliminated, ensuring that CD4+ and CD8+ T cells recognise only foreign antigens presented by self-HLA molecules [48]. However, certain HLA alleles, including RA, can contribute to immune dysregulation and autoimmunity. CD4+ T cells are essential for antibody maturation in germinal centres of lymph nodes, where they interact with B cells and secrete cytokines that promote B cell proliferation, differentiation, and somatic hypermutation. B cell clones with the highest antigen-binding affinity are selected through interactions with follicular dendritic cells, leading to affinity maturation [48, 49].
Genetic factors account for approximately 60% of the risk for developing RA, with the HLA-DRβ chain containing a five-amino-acid sequence motif, known as the shared epitope (SE), a significant risk factor for ACPA-positive RA [18, 50]. A review article by Sharma et al. [51] emphasises significant HLA loci related to RA, which are the HLA class II gene (a.a. position 9 of HLA-DPβ1) and HLA class I genes (a.a. position 9 of HLA-β and position 77 of HLA-α). In addition to HLA regions, non-HLA loci, such as protein tyrosine phosphatase N22 (PTPN22), also contribute to the genetic risk of ACPA-positive RA. Up to 2024, more than 150 loci have been reported to be associated with RA risk. However, whether RA susceptibility is driven by the HLA gene or other associated genes remains unclear, necessitating further research [18, 51].
ACPA-producing germinal centre B cells may escape stringent selection due to the abundance of citrullinated antigens [52]. Activated CD4+ T cells likely support ACPA maturation. Evidence suggests the HLA class II locus is more linked to ACPA+ RA progression than initial ACPA formation, indicating its role in ACPA maturation. This is reflected in isotype expansion, increased ACPA levels, and spreading epitope, emphasising HLA class II’s importance in ACPA development [52, 53].
IC disorder is induced by the soluble antigen-antibody complexes deposition in vessel walls or the basement membrane of the kidneys or by the in-situ development of adherent IC from antibody binding to tissue antigens [41]. The factors influencing the secreted IC to lead to tissue deposition and disease remain unclear, but the properties of IC and local vascular alteration are the key influencing factors. The pathogenicity of IC by depositing on tissues depends on its antigen-antibody ratio [12, 13]. As antibody present in excess, ICs become insoluble, unable to circulate and phagocytosed by macrophages, which is considered pathogenic [10, 12, 13, 24]. Generally, ICs get concentrated and deposited in organs where blood is filtered at high pressure to form other fluids, such as synovial fluids and damage the joints [12, 13].
With increased circulating ICs concentration, the complexes are prone to deposit in tissues and trigger FcR-expressing immune cells [8, 24], initiate neutrophilic inflammation and cause host tissue damage [8, 54, 55]. Furthermore, the deposited ICs in the synovial region significantly mediate neutrophil activation and tissue inflammation [56]. Figure 6 summarises the possible mechanism of synovial inflammation observed in the early onset of RA.
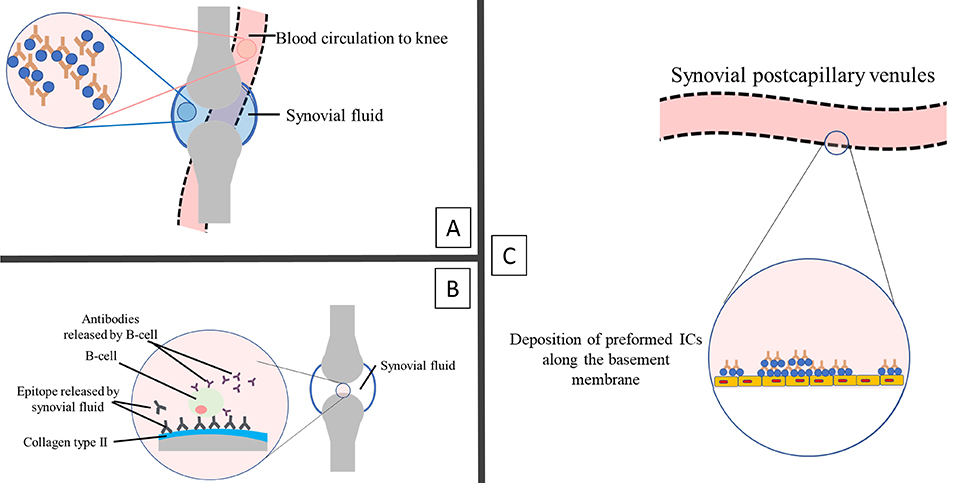
Possible mechanism of early synovial inflammation in rheumatoid arthritis. (A): IC or autoantibodies directly enter the synovium from circulation [57]. (B): Specific antigens located at the cartilage bind directly to the autoantibodies that may reach the synovial pannus tissue [25], as discussed earlier in stage 1: immune system formation. (C): IC deposited along the basement membrane of synovial postcapillary venules from the circulation can initiate vasculitis [58, 59]. The binding of IC to the circulated immune cell influences the release of vasoactive mediators, which causes the leakage of vascular synovial vasculature and subsequently aids the entry of autoantibodies [60]. IC: immune complex
Generally, ICs induce an acute inflammatory response via complement activation and interaction of leukocyte FcR once deposited in tissues [12, 24]. The tissue damage is equivalent regardless of which ICs are deposited. The resultant inflammation lesions that develop in the joints are arthritis [12, 13]. ICs formation, complement fixation, chemotactic fragments release and stimulation of the event cascade trigger innate and adaptive immune cell recruitment and facilitate stromal cell activation in RA pathogenesis [27]. Following FcR and complement receptor engagement, ICs activate various cell types, resulting in different effector actions. FcR is involved in initiating and modulating various immunological responses [61, 62]. Deposition of IC into tissues causes cleavage of complement anaphylatoxins C3a and C5a [63], which subsequently induces granule release from mast cells [64] and recruits the inflammatory cells into the tissue. The lysosomal action of inflammatory cells promotes tissue injury through phagocytosis by macrophages and polymorphonuclear neutrophils [65].
The complement systems, composed of several proteins, play crucial roles in immune processes, such as processing and clearance of circulating ICs, foreign antigen recognition, humoral and cellular immunity modulation, apoptotic and dead cells removal, and involvement in injury resolving and tissue regeneration [66]. In RA, complement system activation mediates inflammation and tissue damage [67]. Table 1 summarises the complement protein associated with RA. As an antibody to ICs in RA, RF appears to be implicated in complement system activation and the generation of chemotactic and inflammatory mediators, resulting in a state that may be maintained and reinitiated. They develop ACPA complexes or other forms of ICs in the synovial cavity, resulting in non-resolving inflammation. Upon this, RF’s second wave of IC formation occurs as an antibody reactive to the initial ICs. These processes are linked to the consumption of complement and the generation of inflammatory mediators [68, 69]. The presence of RF-IgM or RF-IgA enhances the FcR-mediated immune response, promotes complement activation (classical, alternative, and lectin cascade), and affects the functions of ACPA-ICs [68]. The ICs and ACPA activate the same complement system [66], which is remarkably observable in RA patients’ serum, synovial lining, and synovial fluid [67].
Associated proteins in the complement system that play a variety of immunological activities
| Protein | Role | Research findings | References |
|---|---|---|---|
| C3a |
|
| [35, 55, 69, 70] |
| C3b |
|
| [69, 71] |
| C4b |
|
| [69, 72, 73] |
| C5a |
|
| [35, 46, 70, 74, 75] |
ACPA: anti-citrullinated protein antibodies; FcγR: fragment crystallisable gamma receptor; ICs: immune complexes; IL: interleukin; RA: rheumatoid arthritis; RF: rheumatoid factor; TNF-α: tumour necrosis factor-α
In RA events, concentrated macrophages in the synovial lining secrete cytokines and chemokines that induce inflammation, cartilage, and bone destruction [76]. Macrophages promote the progression of the RA condition by increasing the expression of cytokines such as interleukin (IL), tumour necrosis factor-α (TNF-α), chemokines, and matrix metalloproteinases. Secretion of macrophage-induced TNF-α drives most of the articular and extra-articular pathology associated with RA [77]. An article by Nascimento et al. [78], published in 2025, illustrates the role of TNF-α in intensifying inflammation in RA, suggesting the immunosuppression agent as a treatment for the disease. Secretion of macrophage-induced cytokines in RA conditions involves toll-like receptors (TLRs). These receptors identify the pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs) [27]. In the RA synovial tissues, macrophages respond to the concentrated TLR-2 and TLR-4 [79]. Research done in 2025 by Glanville [80] suggests that microparticles carrying the ICs activate TLR-4, activate a potent DAMP stimulus that drives pro-inflammatory cytokines production and sustains synovial inflammation in RA.
Besides TRLs, cytokine production in RA conditions also involves FcR [81]. The presence of abundant ICs stimulates FcR activation and leads to persistent neutrophilic inflammation by activating the neutrophil, which causes tissue injury [8]. Activated neutrophil stimulates the secretion of IL-1β, which drives synovial cells to form chemokines, responsible for more neutrophil recruitment [56]. Neutrophil FcRs ligation effectively activates pro-inflammatory activities such as reactive oxygen species (ROS), degranulation, and cytokine secretion [82, 83]. Stimulation of ROS secretion by neutrophils leads to tissue injury, endothelial dysfunction, and immunoglobulin mutation, which further causes autoantibody secretion [82, 84]. The cytokines secreted by the neutrophils enhanced the resorption of bone in RA. Neutrophils also stimulate expression of the functionally active, membrane-bound receptor activator of nuclear factor kappa-light chain-enhancer of activated B cells (NF-κB) ligand, which leads to an upsurge of NF-κB ligand in synovial fluid of RA patients [85, 86].
IC-activated neutrophils produce a variety of inflammatory mediators, including significant neutrophil chemoattractants like IL-8 and leukotriene B4, which leads to increased neutrophil recruitment and progressive inflammation [87]. In the pathogenesis of RA, activated neutrophils are directly associated with the expression of cytotoxic capacity [86]. It includes the inhibition of chondrocyte proliferation [79] and activation of synoviocyte proliferation [88] and invasion [89], which leads to progressive joint damage. In addition, activities of the inflammatory cell that expels the produced or deposited ICs on cartilage and the IC-activated neutrophil that releases its granule enzyme into the cartilage surface create a harmful condition termed “frustrated phagocytosis” [35].
By integrating recent immunological insights and emphasising current mechanisms of IC-mediated pathology, this review offers an updated framework for understanding and targeting RA progression. ICs play a dominant role in the pathogenesis of RA by acting as key activators. The formation of ICs in circulation also initiates type III hypersensitivity reactions in RA. Autoantibodies such as RF and ACPAs in RA target immunoglobulin antigens or Fc regions to develop ICs. As these complexes deposit in the joint tissue surface, they initiate inflammatory responses and tissue damage. The inflammation is induced by complement activation, while the recruitments of leukocytes, such as macrophages and neutrophils, by complement products and FcR cause further tissue injury by releasing the pro-inflammatory cytokines and chemokines. Consequently, the central pathogenic process in RA is mediated by ICs to induce immunological responses. Continuous IC formation worsens RA chronicity by providing a positive feedback loop, intensifying the arthritic inflammatory process. Thus, this review may assist in further understanding the mechanisms of systemic IC-mediated RA and enable the development of effective therapeutic strategies that improve life quality.
ACPAs: anti-citrullinated protein antibodies
CII: type II collagen
DAMPs: damage-associated molecular patterns
FcR: fragment crystallisable receptor
GPI: glucose-6-phosphate isomerase
HLA: human leukocyte antigen
ICs: immune complexes
IL: interleukin
NF-κB: nuclear factor kappa-light chain-enhancer of activated B cells
RA: rheumatoid arthritis
RF: rheumatoid factor
ROS: reactive oxygen species
TLRs: toll-like receptors
TNF-α: tumour necrosis factor-α
VK: Conceptualization, Investigation, Writing—original draft, Writing—review & editing, Visualization. NAAB: Validation, Writing—review & editing, Supervision. NAS: Conceptualization, Investigation, Writing—original draft, Writing—review & editing, Supervision. All authors read and approved the submitted version.
The authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
This study is funded by the Malaysian Ministry of Higher Education through Fundamental Research Grant Scheme (FRGS) [Grant Number: FRGS/1/2019/WAB11/UNISZA/03/1]. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
© The Author(s) 2025.
Open Exploration maintains a neutral stance on jurisdictional claims in published institutional affiliations and maps. All opinions expressed in this article are the personal views of the author(s) and do not represent the stance of the editorial team or the publisher.
Copyright: © The Author(s) 2025. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

View: 2208
Download: 32
Times Cited: 0