-
71 results in Exploration of Digestive DiseasesSort byMost Downloaded
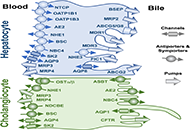 Etiopathogenesis and pathophysiology of cholestasisOpen AccessReviewNormal hepatobiliary function depends on an adequate bile flow from the liver through the biliary tree to the gallbladder, where bile is stored and concentrated, and from the gallbladder to the duod [...] Read more.Maitane Asensio ... Jose J. G. MarinPublished: October 31, 2022 Explor Dig Dis. 2022;1:97–117
Etiopathogenesis and pathophysiology of cholestasisOpen AccessReviewNormal hepatobiliary function depends on an adequate bile flow from the liver through the biliary tree to the gallbladder, where bile is stored and concentrated, and from the gallbladder to the duod [...] Read more.Maitane Asensio ... Jose J. G. MarinPublished: October 31, 2022 Explor Dig Dis. 2022;1:97–117
DOI: https://doi.org/10.37349/edd.2022.00008
This article belongs to the special issue CHOLESTASIS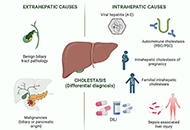 Drug-induced cholestasis: causative agents and challenges in diagnosis and managementOpen AccessReviewDrug-induced liver injury (DILI) is an adverse reaction to drugs and other xenobiotics that can have serious consequences and jeopardise progress in pharmacological therapy. While DILI is predominan [...] Read more.Jose M. Pinazo-Bandera ... Miren García-CortésPublished: September 18, 2023 Explor Dig Dis. 2023;2:202–222
Drug-induced cholestasis: causative agents and challenges in diagnosis and managementOpen AccessReviewDrug-induced liver injury (DILI) is an adverse reaction to drugs and other xenobiotics that can have serious consequences and jeopardise progress in pharmacological therapy. While DILI is predominan [...] Read more.Jose M. Pinazo-Bandera ... Miren García-CortésPublished: September 18, 2023 Explor Dig Dis. 2023;2:202–222
DOI: https://doi.org/10.37349/edd.2023.00027
This article belongs to the special issue CHOLESTASIS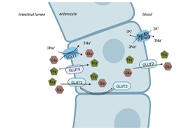 Fructose, a trigger of metabolic diseases?—a narrative reviewOpen AccessReviewWorldwide the number of individuals being overweight or obese has dramatically increased during the last decades, which is also associated with a similar dramatic increase of individuals afflicted w [...] Read more.Anja Baumann ... Ina BergheimPublished: August 29, 2022 Explor Dig Dis. 2022;1:51–71
Fructose, a trigger of metabolic diseases?—a narrative reviewOpen AccessReviewWorldwide the number of individuals being overweight or obese has dramatically increased during the last decades, which is also associated with a similar dramatic increase of individuals afflicted w [...] Read more.Anja Baumann ... Ina BergheimPublished: August 29, 2022 Explor Dig Dis. 2022;1:51–71
DOI: https://doi.org/10.37349/edd.2022.00005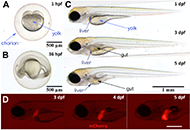 Zebrafish as a model for drug induced liver injury: state of the art and beyondOpen AccessReviewZebrafish as a preclinical drug induced liver injury (DILI) model provides multiple advantages ranging from ease of breeding and maintenance, availability of different strains and transgenic fish am [...] Read more.Gulcin Cakan-Akdogan ... Ozlen KonuPublished: April 26, 2023 Explor Dig Dis. 2023;2:44–55
Zebrafish as a model for drug induced liver injury: state of the art and beyondOpen AccessReviewZebrafish as a preclinical drug induced liver injury (DILI) model provides multiple advantages ranging from ease of breeding and maintenance, availability of different strains and transgenic fish am [...] Read more.Gulcin Cakan-Akdogan ... Ozlen KonuPublished: April 26, 2023 Explor Dig Dis. 2023;2:44–55
DOI: https://doi.org/10.37349/edd.2023.00017
This article belongs to the special issue Drug-induced Liver Injury: From Bench to Clinical Application Helicobacter pylori and gastric cancer: a critical approach to who really needs eradicationOpen AccessReviewIt is generally accepted that eradication of Helicobacter pylori (H. pylori) infection may reduce the risk of the development of gastric cancer. Recommendations for global generalized tests and trea [...] Read more.Elias Kouroumalis ... Argyro VoumvourakiPublished: April 16, 2024 Explor Dig Dis. 2024;3:107–142
Helicobacter pylori and gastric cancer: a critical approach to who really needs eradicationOpen AccessReviewIt is generally accepted that eradication of Helicobacter pylori (H. pylori) infection may reduce the risk of the development of gastric cancer. Recommendations for global generalized tests and trea [...] Read more.Elias Kouroumalis ... Argyro VoumvourakiPublished: April 16, 2024 Explor Dig Dis. 2024;3:107–142
DOI: https://doi.org/10.37349/edd.2024.00043
This article belongs to the special issue Helicobacter Pylori and Infection: Genomics, Diagnosis, Pathogenesis, Antibiotic Resistance, Microbiota, Cancer, Prevention and Therapeutics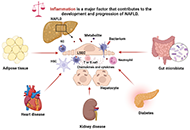 Molecular mechanisms of metabolic disease-associated hepatic inflammation in non-alcoholic fatty liver disease and non-alcoholic steatohepatitisOpen AccessReviewNon-alcoholic fatty liver disease (NAFLD) is the leading chronic liver disease worldwide, with a progressive form of non-alcoholic steatohepatitis (NASH). It may progress to advanced liver diseases, [...] Read more.Chunye Zhang ... Ming YangPublished: October 25, 2023 Explor Dig Dis. 2023;2:246–275
Molecular mechanisms of metabolic disease-associated hepatic inflammation in non-alcoholic fatty liver disease and non-alcoholic steatohepatitisOpen AccessReviewNon-alcoholic fatty liver disease (NAFLD) is the leading chronic liver disease worldwide, with a progressive form of non-alcoholic steatohepatitis (NASH). It may progress to advanced liver diseases, [...] Read more.Chunye Zhang ... Ming YangPublished: October 25, 2023 Explor Dig Dis. 2023;2:246–275
DOI: https://doi.org/10.37349/edd.2023.00029
This article belongs to the special issue Cellular and Molecular Targets for NAFLD or MAFLD Treatments and Their Functions in Liver Fibrosis, Cirrhosis, and Cancer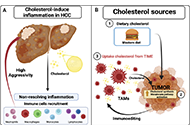 Tumor immune microenvironment modulation by cholesterol in hepatocellular carcinomaOpen AccessReviewHepatocellular carcinoma (HCC) is considered one of the most aggressive tumors worldwide. The consumption of lipid-enriched diets, mainly high cholesterol, induces oxidative stress and chronic infla [...] Read more.Alejandro Escobedo-Calvario ... María Concepción Gutiérrez-RuízPublished: July 29, 2022 Explor Dig Dis. 2022;1:21–39
Tumor immune microenvironment modulation by cholesterol in hepatocellular carcinomaOpen AccessReviewHepatocellular carcinoma (HCC) is considered one of the most aggressive tumors worldwide. The consumption of lipid-enriched diets, mainly high cholesterol, induces oxidative stress and chronic infla [...] Read more.Alejandro Escobedo-Calvario ... María Concepción Gutiérrez-RuízPublished: July 29, 2022 Explor Dig Dis. 2022;1:21–39
DOI: https://doi.org/10.37349/edd.2022.00003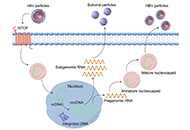 Hepatitis B virus: modes of transmission, immune pathogenesis, and research progress on therapeutic vaccinesOpen AccessReviewHepatitis B virus (HBV) infection affects 262 million people worldwide, leading to over 820,000 deaths annually. The reason HBV has been a persistent issue for decades is that it is a non-cytopathic [...] Read more.Chunzheng Li ... Xianguang YangPublished: October 14, 2024 Explor Dig Dis. 2024;3:443–458
Hepatitis B virus: modes of transmission, immune pathogenesis, and research progress on therapeutic vaccinesOpen AccessReviewHepatitis B virus (HBV) infection affects 262 million people worldwide, leading to over 820,000 deaths annually. The reason HBV has been a persistent issue for decades is that it is a non-cytopathic [...] Read more.Chunzheng Li ... Xianguang YangPublished: October 14, 2024 Explor Dig Dis. 2024;3:443–458
DOI: https://doi.org/10.37349/edd.2024.00060
This article belongs to the special issue Viral Hepatitis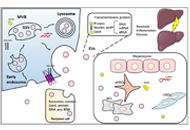 Extracellular vesicles in metabolic dysfunction associated fatty liver disease: mechanisms, diagnostic and therapeutic implicationsOpen AccessReviewThe prevalence of metabolic dysfunction-associated fatty liver disease (MAFLD) is increasing rapidly worldwide due to the obesity epidemic. Advanced stages of the MAFLD, such as non-alcoholic steato [...] Read more.Zongmei Wu ... Han MoshagePublished: July 13, 2022 Explor Dig Dis. 2022;1:4–20
Extracellular vesicles in metabolic dysfunction associated fatty liver disease: mechanisms, diagnostic and therapeutic implicationsOpen AccessReviewThe prevalence of metabolic dysfunction-associated fatty liver disease (MAFLD) is increasing rapidly worldwide due to the obesity epidemic. Advanced stages of the MAFLD, such as non-alcoholic steato [...] Read more.Zongmei Wu ... Han MoshagePublished: July 13, 2022 Explor Dig Dis. 2022;1:4–20
DOI: https://doi.org/10.37349/edd.2022.00002 Diagnostic workup of suspected hereditary cholestasis in adults: a case reportOpen AccessCase ReportHereditary cholestasis comprises a broad spectrum of clinical phenotypes of varying severity. Severe forms such as progressive familial intrahepatic cholestasis (PFIC) mostly affect children with di [...] Read more.Carola Dröge ... Verena KeitelPublished: April 21, 2023 Explor Dig Dis. 2023;2:34–43
Diagnostic workup of suspected hereditary cholestasis in adults: a case reportOpen AccessCase ReportHereditary cholestasis comprises a broad spectrum of clinical phenotypes of varying severity. Severe forms such as progressive familial intrahepatic cholestasis (PFIC) mostly affect children with di [...] Read more.Carola Dröge ... Verena KeitelPublished: April 21, 2023 Explor Dig Dis. 2023;2:34–43
DOI: https://doi.org/10.37349/edd.2023.00016
This article belongs to the special issue CHOLESTASIS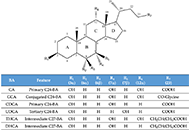 Cholestasis associated to inborn errors in bile acid synthesisOpen AccessReviewSeveral metabolic pathways are involved in the biotransformation of C27 neutral cholesterol to C24 primary bile acids (BAs), mainly cholic acid (CA) and chenodeoxycholic acid (CDCA), which are then conjugated with glycine or tauri [...] Read more.Ricardo Espinosa-Escudero ... Maria J. MontePublished: December 7, 2022 Explor Dig Dis. 2022;1:137–153
Cholestasis associated to inborn errors in bile acid synthesisOpen AccessReviewSeveral metabolic pathways are involved in the biotransformation of C27 neutral cholesterol to C24 primary bile acids (BAs), mainly cholic acid (CA) and chenodeoxycholic acid (CDCA), which are then conjugated with glycine or tauri [...] Read more.Ricardo Espinosa-Escudero ... Maria J. MontePublished: December 7, 2022 Explor Dig Dis. 2022;1:137–153
DOI: https://doi.org/10.37349/edd.2022.00010
This article belongs to the special issue CHOLESTASIS Alcohol-related liver disease: also a question of what you drink?Open AccessReviewExcessive alcohol intake is still among the leading causes of chronic liver diseases. Epidemiological studies suggest that per capita consumption of alcohol from various alcohol beverages e.g., beer [...] Read more.Finn Jung ... Ina BergheimPublished: June 30, 2023 Explor Dig Dis. 2023;2:118–132
Alcohol-related liver disease: also a question of what you drink?Open AccessReviewExcessive alcohol intake is still among the leading causes of chronic liver diseases. Epidemiological studies suggest that per capita consumption of alcohol from various alcohol beverages e.g., beer [...] Read more.Finn Jung ... Ina BergheimPublished: June 30, 2023 Explor Dig Dis. 2023;2:118–132
DOI: https://doi.org/10.37349/edd.2023.00022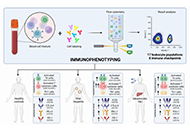 Immunophenotyping to improve the mechanistic understanding of idiosyncratic drug-induced liver injury: clinical implications and future directionsOpen AccessReviewThe late event onset of a fraction of idiosyncratic drug-induced liver injury (DILI) cases and the link observed by genome-wide association studies (GWASs) of certain human leucocyte antigen (HLA) a [...] Read more.Alejandro Cueto-Sánchez ... Marina Villanueva-PazPublished: April 26, 2023 Explor Dig Dis. 2023;2:56–76
Immunophenotyping to improve the mechanistic understanding of idiosyncratic drug-induced liver injury: clinical implications and future directionsOpen AccessReviewThe late event onset of a fraction of idiosyncratic drug-induced liver injury (DILI) cases and the link observed by genome-wide association studies (GWASs) of certain human leucocyte antigen (HLA) a [...] Read more.Alejandro Cueto-Sánchez ... Marina Villanueva-PazPublished: April 26, 2023 Explor Dig Dis. 2023;2:56–76
DOI: https://doi.org/10.37349/edd.2023.00018
This article belongs to the special issue Drug-induced Liver Injury: From Bench to Clinical Application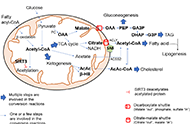 The central role of mitochondrial metabolism in hepatic steatosisOpen AccessReviewMitochondria are present in all mammalian cells except matured red blood cells. Mitochondria consist of several metabolic pathways for glucose, fatty acids, amino acids, and bioenergetic pathways fo [...] Read more.Sanda Win ... Filbert Win Min AungPublished: February 29, 2024 Explor Dig Dis. 2024;3:42–68
The central role of mitochondrial metabolism in hepatic steatosisOpen AccessReviewMitochondria are present in all mammalian cells except matured red blood cells. Mitochondria consist of several metabolic pathways for glucose, fatty acids, amino acids, and bioenergetic pathways fo [...] Read more.Sanda Win ... Filbert Win Min AungPublished: February 29, 2024 Explor Dig Dis. 2024;3:42–68
DOI: https://doi.org/10.37349/edd.2024.00039
This article belongs to the special issue Mitochondria and Lipid Signalling in Liver Diseases Hepatocellular carcinoma surveillance after HBsAg seroclearanceOpen AccessReviewHepatitis B surface antigen (HBsAg) seroclearance is considered the functional cure and the optimal treatment endpoint for chronic hepatitis B (CHB). Patients with CHB who cleared HBsAg generally ha [...] Read more.Jimmy Che-To Lai ... Terry Cheuk-Fung YipPublished: May 11, 2024 Explor Dig Dis. 2024;3:175–189
Hepatocellular carcinoma surveillance after HBsAg seroclearanceOpen AccessReviewHepatitis B surface antigen (HBsAg) seroclearance is considered the functional cure and the optimal treatment endpoint for chronic hepatitis B (CHB). Patients with CHB who cleared HBsAg generally ha [...] Read more.Jimmy Che-To Lai ... Terry Cheuk-Fung YipPublished: May 11, 2024 Explor Dig Dis. 2024;3:175–189
DOI: https://doi.org/10.37349/edd.2024.00046
This article belongs to the special issue Chronic Hepatitis B and C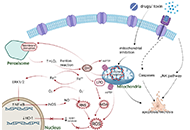 Role of oxidative stress and endoplasmic reticulum stress in drug-induced liver injuryOpen AccessReviewThe pathogenesis of drug-induced liver injury (DILI) is still in an early stage of research. However, investigators have shown that both oxidative stress and endoplasmic reticulum (ER) stress play a [...] Read more.Hanghang Wu ... Francisco Javier CuberoPublished: June 28, 2023 Explor Dig Dis. 2023;2:83–99
Role of oxidative stress and endoplasmic reticulum stress in drug-induced liver injuryOpen AccessReviewThe pathogenesis of drug-induced liver injury (DILI) is still in an early stage of research. However, investigators have shown that both oxidative stress and endoplasmic reticulum (ER) stress play a [...] Read more.Hanghang Wu ... Francisco Javier CuberoPublished: June 28, 2023 Explor Dig Dis. 2023;2:83–99
DOI: https://doi.org/10.37349/edd.2023.00020
This article belongs to the special issue Drug-induced Liver Injury: From Bench to Clinical Application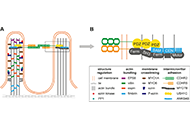 The intermicrovillar adhesion complex in gut barrier function and inflammationOpen AccessPerspectiveThe surface of intestinal epithelial cells is covered by the brush border, which consists of densely packed cellular extrusions called microvilli. Until recently, microvilli have not been known to b [...] Read more.Bernadette Mödl ... Robert EferlPublished: October 11, 2022 Explor Dig Dis. 2022;1:72–79
The intermicrovillar adhesion complex in gut barrier function and inflammationOpen AccessPerspectiveThe surface of intestinal epithelial cells is covered by the brush border, which consists of densely packed cellular extrusions called microvilli. Until recently, microvilli have not been known to b [...] Read more.Bernadette Mödl ... Robert EferlPublished: October 11, 2022 Explor Dig Dis. 2022;1:72–79
DOI: https://doi.org/10.37349/edd.2022.00006 The hepatocyte growth factor induces an anti-inflammatory and repairing response in the cholestasis-induced colon damageOpen AccessOriginal ArticleAim: Cholestasis remains a partially characterized disease. Evidence has been gained that it is a systemic disease that begins in the liver but significantly impacts other organs and systems such [...] Read more.Jocelyn López-Ramirez ... Leticia Bucio-OrtizPublished: August 11, 2022 Explor Dig Dis. 2022;1:40–50
The hepatocyte growth factor induces an anti-inflammatory and repairing response in the cholestasis-induced colon damageOpen AccessOriginal ArticleAim: Cholestasis remains a partially characterized disease. Evidence has been gained that it is a systemic disease that begins in the liver but significantly impacts other organs and systems such [...] Read more.Jocelyn López-Ramirez ... Leticia Bucio-OrtizPublished: August 11, 2022 Explor Dig Dis. 2022;1:40–50
DOI: https://doi.org/10.37349/edd.2022.00004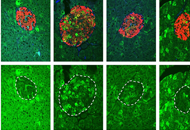 Probiotic human alcohol dehydrogenase-4 expressing bacteria protects from diet-induced obesity and metabolic impairment: a new concept of disease preventionOpen AccessOriginal ArticleAim: Probiotic bacteria consumption for improving human health and for disease prevention is still controversial. There is a need to develop functional probiotic bacteria with proven efficacy for [...] Read more.Rajnish Prakash Singh ... Zvi HayoukaPublished: October 31, 2022 Explor Dig Dis. 2022;1:118–136
Probiotic human alcohol dehydrogenase-4 expressing bacteria protects from diet-induced obesity and metabolic impairment: a new concept of disease preventionOpen AccessOriginal ArticleAim: Probiotic bacteria consumption for improving human health and for disease prevention is still controversial. There is a need to develop functional probiotic bacteria with proven efficacy for [...] Read more.Rajnish Prakash Singh ... Zvi HayoukaPublished: October 31, 2022 Explor Dig Dis. 2022;1:118–136
DOI: https://doi.org/10.37349/edd.2022.00009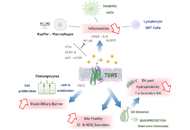 The bile acid receptor TGR5 and cholestasisOpen AccessReviewMetabolic zonation in the liver carries out the maintenance of organ and body homeostasis. Hypoxia is an inherent physiological feature of the liver and contributes to the zonal properties of the he [...] Read more.Grégory Merlen ... Thierry TordjmannPublished: December 30, 2022 Explor Dig Dis. 2022;1:154–169
The bile acid receptor TGR5 and cholestasisOpen AccessReviewMetabolic zonation in the liver carries out the maintenance of organ and body homeostasis. Hypoxia is an inherent physiological feature of the liver and contributes to the zonal properties of the he [...] Read more.Grégory Merlen ... Thierry TordjmannPublished: December 30, 2022 Explor Dig Dis. 2022;1:154–169
DOI: https://doi.org/10.37349/edd.2022.00011
This article belongs to the special issue CHOLESTASIS -
