Original Article
Original Article

Affiliation:
1Division of Applied Biomedical Sciences and Biotechnology, IMU University, Bukit Jalil, 57000 Kuala Lumpur, Malaysia
ORCID: https://orcid.org/0000-0001-5252-0516
Affiliation:
2School of Pharmacy, Monash University Malaysia, 4700 Bandar Sunway, Selangor, Malaysia
ORCID: https://orcid.org/0000-0002-5164-1970
Affiliation:
3Department of Biomedical Science, University of Nottingham Malaysia Campus, 43500 Semenyih, Selangor, Malaysia
ORCID: https://orcid.org/0000-0002-4546-2351
Affiliation:
4Jeffrey Cheah School of Medicine and Health Sciences, Monash University Malaysia, 47500 Bandar Sunway, Selangor, Malaysia
ORCID: https://orcid.org/0000-0002-8615-8241
Affiliation:
5School of Pharmacy, IMU University, Bukit Jalil, 57000 Kuala Lumpur, Malaysia
Email: ceong98@hotmail.com
ORCID: https://orcid.org/0000-0001-5926-7250
Explor Drug Sci. 2025;3:1008111 DOl: https://doi.org/10.37349/eds.2025.1008111
Received: March 14, 2025 Accepted: May 01, 2025 Published: May 27, 2025
Academic Editor: Prasat Kittakoop, Chulabhorn Graduate Institute, Thailand
Aim: This study aimed to elucidate the structural basis for the interaction of five natural anti-arthritic compounds, diacerein, rhein, glucosamines [glucosamine 3-sulfate (G3S), and glucosamine 6-sulfate (G6S)], and chondroitin disaccharide Δdi-4S (C4S) with cytochrome P450 2C9 (CYP2C9).
Methods: The investigated compounds were docked individually to the defined binding site in CYP2C9 based on the published crystal structure (PDB code: 1R9O).
Results: All investigated ligands bound deep in the active site pocket in close proximity to the heme. Except for chondroitin, all ligands are bonded to residues found in critical secondary structures that form the boundary of the active site cavity, including B-C loop, F helix, F-G loop, and I helix. A total of 12 amino acids were involved in the binding, and all were critical residues located in four out of six substrate recognition sites (SRSs) that have been identified as important substrate binding and catalysis regions in other CYP isoforms. The relatively more potent binding (lower CDOCKER interaction energy) observed for diacerein and rhein compared to glucosamines and C4S are likely due to two main factors: a higher number of bonds between the ligand molecule and CYP2C9 active site residues (14 versus 0–4), and direct interaction with the heme moiety. The binding residues identified in both diacerein and rhein were the residues that also bonded with sulfaphenazole, the specific and potent CYP2C9 inhibitor.
Conclusions: Collectively, this study has provided insights into structural features of CYP2C9 critical for inhibition and formed a basis for further exploration of structural determinants for potency and specificity of therapeutic compounds as CYP2C9 inhibitors.
One of the three human microsomal CYPs in subfamily 2C that significantly contribute to the hepatic metabolism of clinically used drugs is cytochrome P450 2C9 (CYP2C9). Antidepressant amitriptyline, antihypertensive losartan, anticancer cyclophosphamide, antidiabetic gliclazide, and nonsteroidal anti-inflammatory drugs like celecoxib and diclofenac are examples of CYP2C9 substrates. Some of the substrates, like phenytoin and warfarin, have a narrow therapeutic index [1]. Numerous therapeutic agents, including the antifungal fluconazole, the antiarrhythmic amiodarone, the antibacterial sulfaphenazole, and the anti-inflammatory phenylbutazone, also suppress CYP2C9 activity. When a patient takes CYP2C9 substrates along with inducers or inhibitors, drug-drug interactions may occur. For instance, people using nonsteroidal anti-inflammatory drugs and warfarin have been known to experience bleeding in their gastrointestinal tracts as a result of the latter’s excessive anticoagulant activity brought on by decreased metabolism [2].
Osteoarthritis (OA), also known as degenerative arthritis or degenerative joint disease, is a common, chronic disease that affects about 15% of the world’s population [3]. Glucosamine, chondroitin, and diacerein are three naturally derived compounds used in the treatment of OA. Glucosamine and chondroitin, both natural constituents of the cartilaginous matrix, are commonly available as dietary supplements, or nutraceuticals [4]. Diacerein is a symptomatic, slow-acting drug for OA. It is an anthraquinone compound of vegetal origin, obtained from the extraction products of aloe or senna leaves. Prior to entering systemic circulation, diacerein is entirely converted to its deacetylated active metabolite, rhein, in the liver [5]. Several studies on the efficacy and safety of diacerein in the treatment of OA have suggested the symptom- and structure-modifying effects of this compound in patients with OA [6, 7].
Despite their widespread use in the treatment of OA, research on the ability of glucosamine, chondroitin, or diacerein to modulate CYP enzymes in humans is relatively scarce. Only a handful of studies have been carried out to date in investigating the modulatory effects of these compounds on CYP enzymatic activity. Persiani and colleagues, in an in vitro study using cryopreserved human hepatocytes and recombinant human CYPs, did not find crystalline glucosamine sulfate to be able to induce or inhibit the catalytic activity of the tested CYPs up to a concentration of 3 mM, the concentration which was hundred folds higher than peak plasma concentrations observed in humans (~10 μM) [8]. Another study demonstrated that chronic administration of chondroitin sulfate did not affect the protein expression and activity of rabbit CYP1A2 and CYP3A6, both in vivo and in vitro [9]. A study by Yokotani et al. [10] has found that both chondroitin sulfate and glucosamine sulfate did not induce or inhibit mice hepatic CYPs, including CYP1A1, CYP1A2, CYP2B, CYP2C, and CYP3A. The same study also suggested no interactions of these two compounds with the anticoagulation activity of warfarin in mice models. An in vitro study using rhein, the major diacerein metabolite, has detected different degrees of inhibition on several CYP isoforms in rats, with the inhibition potency in the following order: CYP2E1 > CYP3A > CYP2C9 > CYP1A2 > CYP2D6 [11]. Based on the inhibition of recombinant human CYP3A4 and CYP2D6 by the ethanol extracts from Aloe vera juice, another study has proposed the involvement of rhein (a constituent of Aloe vera, although its concentration in the juice extracts was not determined) in inhibiting the activity of these two CYP isoforms [12].
We have previously published a study investigating the inhibitory magnitude of glucosamine, chondroitin, diacerein, and rhein on CYP2C9 using an in vitro inhibition assay and molecular docking [13]. Utilizing valsartan hydroxylase assay as probe, all forms of glucosamine and chondroitin exhibited negligible inhibition toward CYP2C9, whereas diacerein and rhein showed moderate to relatively strong degree of inhibition. The in silico data from the study correlated well with in vitro experimental data, where both diacerein and rhein have shown lower binding free energy than glucosamine and chondroitin, with interactions dominated by hydrogen and hydrophobic bondings. In the present study, we have expanded the docking simulation to include elucidation of residue interaction, critical residues responsible for inhibitor binding, as well as the nature of the bonding interactions. The obtained results have provided insights into structural features of CYP2C9 critical for inhibition at the atomic level and may form the basis for future research in designing anti-arthritic compounds with reduced drug interaction or inhibition liability.
The RCSB protein data bank (http://www.pdb.org) provided the PDB file for the crystal structure of human CYP2C9 complexed with flurbiprofen (EC: 1.14.13, 1R9O.pdb). The CDOCKER protocol for receptor-ligand interactions section of Discovery Studio 4.5 (DS 4.5) (Accelrys Software Inc., San Diego, USA) was used to carry out the molecular docking process. This is an implementation of a CHARMm based molecular docking tool using a rigid receptor [14]. The CDOCKER algorithm employs a methodology that generates many initial ligand orientations in the target protein’s active site, followed by simulated annealing based on molecular dynamics (MD) and energy minimization for final refinement. Using the replica feature of the CHARMm package, the annealing schedule is implemented as a set of scripts that engage a sequence of heating and cooling steps.
At first, the enzyme and the ligands were both pretreated. All the anti-arthritic compounds’ 3D structures were created using ChemBioDraw Ultra 13.0 (CambridgeSoft, Perkin Elmer, USA), and the AM1 approach was used to optimize them. In order to prepare the enzyme, hydrogen atoms were added while the protein’s pH was adjusted between 6.5 and 8.5. DS 4.5 was subsequently used to eliminate the water molecules from the crystal structure and to charge the ferrous ion in the heme to match the physiological state. CHARMm was finally used to refine the protein. Using DS 4.5, the active site of CYP2C9 was automatically searched and chosen based on relevant references. Using various random seeds and high temperature MD, a number of ligand conformations were produced. By shifting the ligand’s center to a predetermined location inside the receptor active site and performing a sequence of arbitrary rotations, random orientations of the conformations were produced. When the softened energy was less than a predetermined threshold, the orientation was maintained. Until the target number of low-energy orientations was achieved or the maximum number of test times for negative orientations was reached, this process was repeated. MD simulated annealing was applied to each orientation. After reaching a high temperature, the temperature was lowered to the target level. Using a non-softened potential, the ligand’s ultimate energy was minimized in the rigid receptor. The interaction energy alone and the CHARMm energy (interaction energy plus ligand strain) were calculated for each of the final poses. The poses that scored the highest (most negative, so favorable to binding) were kept after the poses were classified based on CHARMm energy. The types of interactions between the docked enzyme and ligand were examined following the completion of molecular docking. Each ligand’s dock score function was used to rank the ten best molecular docking poses. The most appropriate pose was determined to be the one with the lowest CDOCKER interaction energy. During the analysis, only amino acid residues and ligands that were within 6.0 Å were permitted. A docking was deemed unsuccessful if it had no output pose. Both 2D and 3D representations of each pose were shown. The CDOCKER protocol’s 2D diagram function for receptor-ligand interactions was used to create the 2D representations.
For the ligand docking simulation, the structure of human CYP2C9 co-crystallized with flurbiprofen (PDB code: 1R9O) was chosen as the protein template. As previously reported, the model has been validated and energy minimized [13]. For the docked flurbiprofen (the docked ligand), the model showed a root mean square deviation (RMSD) value of 1.8 Å, which was well below the cut-off value of 2 [15]. This suggests that our docking process was adequately validated and that the reproduction quality was acceptable. The energy-optimized sulfaphenazole, diacerein, rhein, glucosamines [both glucosamine 3-sulfate (G3S) and G6S], and chondroitin disaccharide Δdi-4S (C4S) were docked individually to the defined binding site in CYP2C9. Figure 1 shows the 3D and 2D representations of the docking of each ligand within CYP2C9 binding site. The pose with the lowest CDOCKER interaction energy was selected as the most favorable pose for each ligand. Figure 2 illustrates the superimposed structures for all investigated compounds; all ligands were interacted at a similar binding region and accommodated the same hydrophobic cleft as flurbiprofen (in the original crystallographic structure) and bonded in close proximity to the heme. The number and type of bonding between ligand molecules and CYP2C9 binding cavity residues, the bond distance, together with the binding interaction energy and IC50 values from the in vitro inhibition assay reported previously [13], are listed in Table 1. The number of bonds displayed in the table differs from that of the earlier study [13] because we aimed to illustrate the full details of the bonding for each residue involved whereas in the previous study, the bonding was reported based on the residues per se without taking into account the number of bonding for each residue.
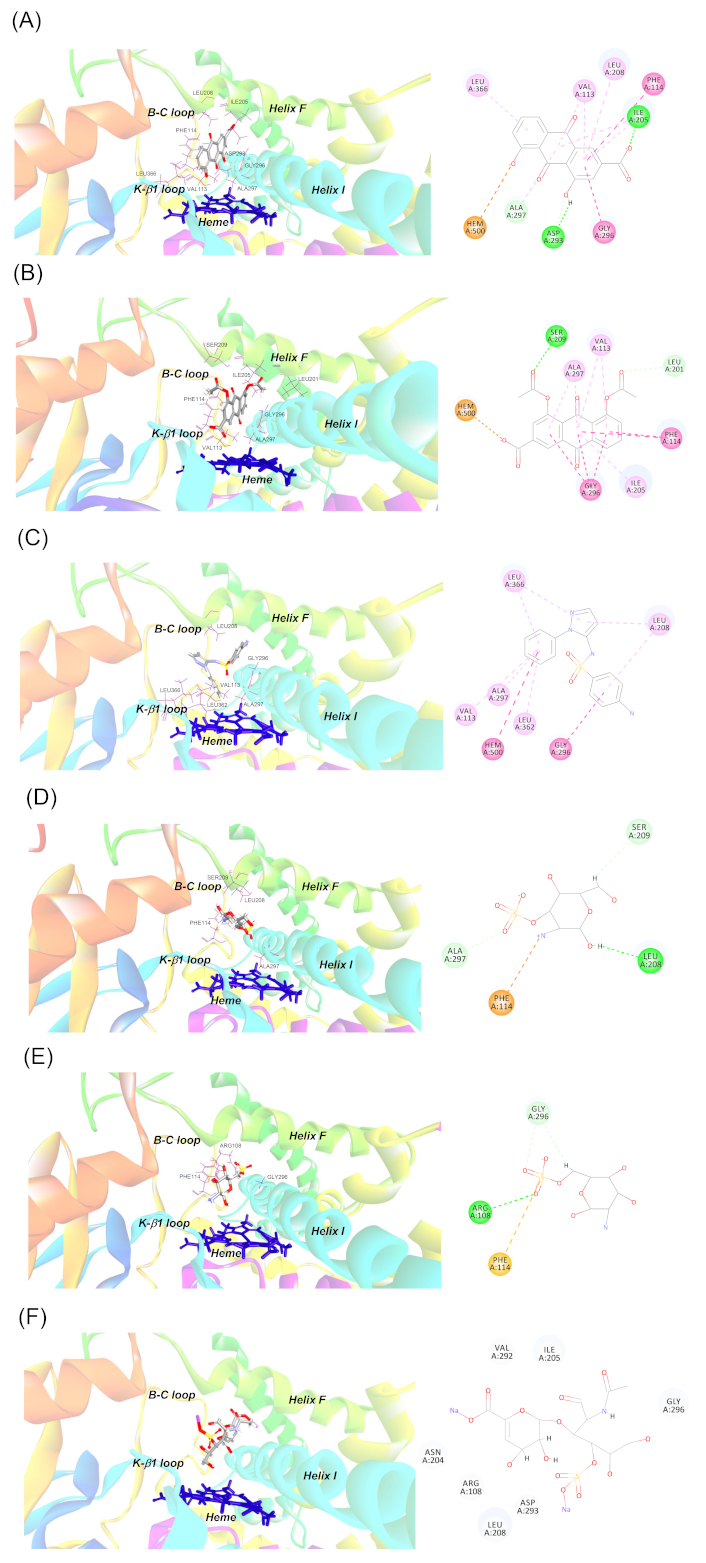
Binding mode of (A) rhein, (B) diacerein, (C) sulfaphenazole, (D) glucosamine 3-sulfate, (E) glucosamine 6-sulfate, and (F) chondroitin disaccharide Δdi-4S to the active site of CYP2C9. Diagrams on the left panel show the 3D representations of the best docked poses of the ligand molecules within CYP2C9 active site, where the protein residues are shown as stick models (as labelled), the ligands are shown as ball and stick models, and the heme group is shown in dark blue. The CYP2C9 secondary structures lining the active site boundary are also displayed and labelled. The diagrams on the right panel show the 2D interactions of the ligands with CYP2C9 active site residues. The dashed lines represent the different types of bonds in various colors: conventional hydrogen bond (green), carbon-hydrogen bond (cyan), pi-pi or amide-pi bond (purple), pi-alkyl bond (light purple), pi-anion or pi-cation bond (orange), and pi-sulfur bond (light brown)
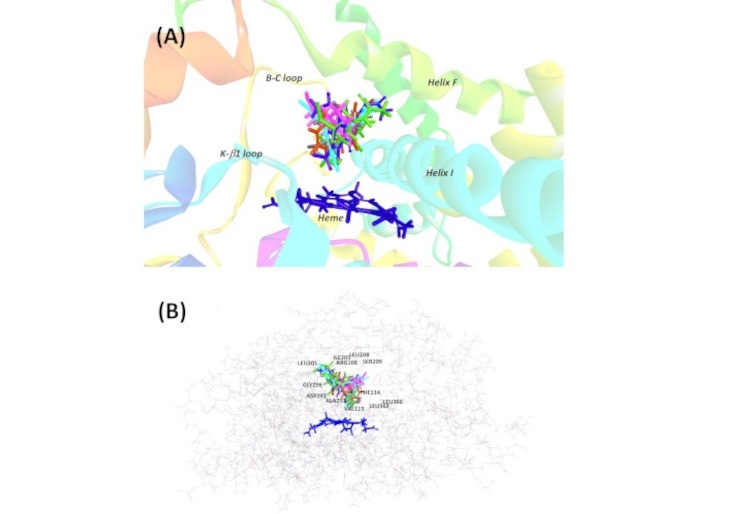
Superimposed image of all investigated ligands within the active site of CYP2C9. (A) The ligands are differentiated with different colors: rhein (brown), diacerein (blue), sulfaphenazole (cyan), glucosamine 3-sulfate (pink), glucosamine 6-sulfate (grey), and chondroitin disaccharide Δdi-4S (green). (B) The binding pocket surrounded with ligands is depicted in the wire style of protein along with the active site residues
The number and type of bonding between ligand molecules and CYP2C9 binding cavity residues, the bond distance, together with the binding interaction energy and IC50 values from the in vitro inhibition assay
| CYP-ligand(Binding energy, kcal/mol; IC50, μM)a | No of bonds | Bonding no | Residues involvedb | Bonding category | Type of bonding | Distance (Å) |
|---|---|---|---|---|---|---|
| CYP2C9-rhein (–35.22; 6.08) | 14 | 1 | A:ILE205:HN1 - Rhein:O17 | Hydrogen Bond | Conventional Hydrogen Bond | 2.562197 |
| 2 | Rhein:H27 - A:ASP293:O | Hydrogen Bond | Conventional Hydrogen Bond | 3.090060 | ||
| 3 | A:ASP293:HA - Rhein:O21 | Hydrogen Bond | Carbon Hydrogen Bond | 2.804421 | ||
| 4 | A:ALA297:HA - Rhein:O18 | Hydrogen Bond | Carbon Hydrogen Bond | 2.870330 | ||
| 5 | Rhein:O20 - A:HEM500 | Electrostatic | Pi-Anion | 4.453505 | ||
| 6 | A:PHE114 - Rhein (ring C) | Hydrophobic | Pi-Pi T-shaped | 5.281925 | ||
| 7 | A:GLY296:C,O; ALA297:N - Rhein (ring C) | Hydrophobic | Amide-Pi Stacked | 4.010072 | ||
| 8 | Rhein (ring C) - A:VAL113 | Hydrophobic | Pi-Alkyl | 5.321472 | ||
| 9 | Rhein (ring C) - A:ILE205 | Hydrophobic | Pi-Alkyl | 5.113695 | ||
| 10 | Rhein (ring C) - A:LEU208 | Hydrophobic | Pi-Alkyl | 5.201962 | ||
| 11 | Rhein (ring B) - A:VAL113 | Hydrophobic | Pi-Alkyl | 5.146036 | ||
| 12 | Rhein (ring B) - A:ILE205 | Hydrophobic | Pi-Alkyl | 5.318006 | ||
| 13 | Rhein (ring B) - A:ALA297 | Hydrophobic | Pi-Alkyl | 5.331537 | ||
| 14 | Rhein (ring A) - A:LEU366 | Hydrophobic | Pi-Alkyl | 4.682293 | ||
| CYP2C9-diacerein (–39.86; 32.23) | 14 | 1 | A:SER209:HN1 - Diacerein:O26 | Hydrogen Bond | Conventional Hydrogen Bond | 2.903369 |
| 2 | A:SER209:HA - Diacerein:O26 | Hydrogen Bond | Conventional Hydrogen Bond | 2.612144 | ||
| 3 | A:LEU201:HA - Diacerein:O22 | Hydrogen Bond | Carbon Hydrogen Bond | 2.743063 | ||
| 4 | Diacerein:O17 - A:HEM500 | Electrostatic | Pi-Anion | 4.831412 | ||
| 5 | A:PHE114 - Diacerein (ring B) | Hydrophobic | Pi-Pi T-shaped | 5.611757 | ||
| 6 | A:PHE114 - Diacerein (ring A) | Hydrophobic | Pi-Pi T-shaped | 4.960882 | ||
| 7 | A:GLY296:C,O; ALA297:N - Diacerein (ring C) | Hydrophobic | Amide-Pi Stacked | 4.812890 | ||
| 8 | A:GLY296:C,O; ALA297:N - Diacerein (ring B) | Hydrophobic | Amide-Pi Stacked | 3.823132 | ||
| 9 | A:GLY296:C,O; ALA297:N - Diacerein (ring A) | Hydrophobic | Amide-Pi Stacked | 4.320433 | ||
| 10 | Diacerein (ring C) - A:ALA297 | Hydrophobic | Pi-Alkyl | 4.973498 | ||
| 11 | Diacerein (ring B) - A:VAL113 | Hydrophobic | Pi-Alkyl | 4.854490 | ||
| 12 | Diacerein (ring B) - A:ILE205 | Hydrophobic | Pi-Alkyl | 5.201749 | ||
| 13 | Diacerein (ring B) - A:ALA297 | Hydrophobic | Pi-Alkyl | 5.027617 | ||
| 14 | Diacerein (ring A) - A:VAL113 | Hydrophobic | Pi-Alkyl | 5.227213 | ||
| CYP2C9-sulfaphenazole (–36.50; 0.95) | 9 | 1 | A:HEM500 - Sulfaphenazole | Hydrophobic | Pi-Pi T-shaped | 5.566038 |
| 2 | A:GLY296:C,O; ALA297:N - Sulfaphenazole (ring A) | Hydrophobic | Amide-Pi Stacked | 4.557513 | ||
| 3 | Sulfaphenazole (ring A) - A:LEU208 | Hydrophobic | Pi-Alkyl | 5.469036 | ||
| 4 | Sulfaphenazole (ring B) - A:LEU208 | Hydrophobic | Pi-Alkyl | 4.861211 | ||
| 5 | Sulfaphenazole (ring B) - A:LEU366 | Hydrophobic | Pi-Alkyl | 5.116724 | ||
| 6 | Sulfaphenazole (ring C) - A:VAL113 | Hydrophobic | Pi-Alkyl | 5.492285 | ||
| 7 | Sulfaphenazole (ring C) - A:ALA297 | Hydrophobic | Pi-Alkyl | 4.923605 | ||
| 8 | Sulfaphenazole (ring C) - A:LEU362 | Hydrophobic | Pi-Alkyl | 5.275490 | ||
| 9 | Sulfaphenazole (ring C) - A:LEU366 | Hydrophobic | Pi-Alkyl | 4.735068 | ||
| CYP2C9-glucosamine 3 sulfate (–25.54; > 1,000) | 4 | 1 | Glucosamine 3-sulphate:H24 - A:LEU208:O | Hydrogen Bond | Conventional Hydrogen Bond | 2.158468 |
| 2 | A:ALA297:HA - Glucosamine 3-sulphate:O21 | Hydrogen Bond | Carbon Hydrogen Bond | 2.379240 | ||
| 3 | Glucosamine 3-sulphate:H18 - A:SER209:OG | Hydrogen Bond | Carbon Hydrogen Bond | 2.714800 | ||
| 4 | Glucosamine 3-sulphate:N12 - A:PHE114 | Electrostatic | Pi-Cation | 3.699830 | ||
| CYP2C9-glucosamine 6 sulfate(–24.41; > 1,000) | 4 | 1 | A:ARG108:HH22 - Glucosamine 6-sulphate:O21 | Hydrogen Bond | Conventional Hydrogen Bond | 3.028392 |
| 2 | A:GLY296:HA2 - Glucosamine 6-sulphate:O22 | Hydrogen Bond | Carbon Hydrogen Bond | 2.743366 | ||
| 3 | Glucosamine 6-sulphate:H18 - A:GLY296:O | Hydrogen Bond | Carbon Hydrogen Bond | 2.573529 | ||
| 4 | Glucosamine 6-sulphate:S20 - A:PHE114 | Other | Pi-Sulfur | 5.314108 | ||
| CYP2C9-chondroitin disaccharide Δdi-4S(–7.59; > 1,000) | 0 | - | - | - | - |
The investigated ligands showed the IC50 values in the increasing order of sulfaphenazole < rhein < diacerein < G3S, G6S, and C4S. This was generally in broad agreement with the rank order of binding affinity, based on CDOCKER interaction energy values, that was sulfaphenazole/diacerein/rhein < glucosamines and chondroitin. Generally, all ligands (except for C4S) were docked and orientated in close proximity to the heme moiety with interactions dominated by hydrogen and hydrophobic bonds. A total of 12 active site amino acids (Table 1 and Figure 2) were involved in the binding, namely, ARG108, VAL113, PHE114, LEU201, ILE205, LEU208, SER209, ASP293, GLY296, ALA297, LEU362, and LEU366. The number of bonds formed between ligands and CYP2C9 decreased from rhein (14 bonds) to C4S (0 bond) as listed in Table 1. Both hydrogen and hydrophobic bonds were featured prominently in CYP2C9-ligand interaction with hydrophobic bonding in the form of pi-pi T-shaped, pi-alkyl, amide-pi stacked interactions observed. In addition, electrostatic bondings involving pi-anion and pi-cation interactions were noted in rhein, diacerein, and G3S. Pi-sulfur bonding was also noted in G6S.
The docking data from this study indicate that all ligands accommodated the same hydrophobic cleft as flurbiprofen (in the original crystallographic structure), and bonded in close proximity to the heme (Figure 2). The number and type of bonding between ligand molecules and CYP2C9 binding cavity residues differed from one ligand to another, resulting in differential binding poses and affinity.
As reported in our previous study [13] and listed in Table 1, sulfaphenazole showed the lowest IC50 value (0.95 μM) toward valsartan-4 hydroxylase activity, confirming its status as the specific and potent inhibitor probe for CYP2C9. Rhein and diacerein were the next potent inhibitors demonstrating IC50 values of 6.08 μM and 32.23 μM respectively. G3S, G6S, and C4S were found to have IC50 values higher than 1,000 μM, indicating negligible inhibition toward CYP2C9. The rank order of binding affinity, based on CDOCKER interaction energy values, was sulfaphenazole/diacerein/rhein > glucosamines and chondroitin. Except for C4S, all ligands were docked and orientated in close proximity to the heme moiety with interactions dominated by hydrogen and hydrophobic bondings. Binding of the docked ligands involved a total of 12 active site amino acids. The majority of these residues possess hydrophobic or non-polar side chains (VAL113, PHE114, LEU201, ILE205, LEU208, GLY296, ALA297, LEU362, and LEU366), only one carries neutral (uncharged) side chain (SER209) and another two have polar side chains (ARG108 and ASP293). All these twelve residues have been found forming the boundary of the active site cavity of CYP2C9 crystal structure [16]. Of these residues, ARG108, VAL113, PHE114, ILE205, LEU208, ASP293, GLY296, ALA297, and LEU366 were shown to reside within 5 Å of flurbiprofen, the co-crystalized ligand in the crystal structure, indicating their primary roles in ligand binding. All the 12 residues in Table 1 were also residues identified within the substrate recognition sites (SRSs) which have been identified by Gotoh [17] as regions important for CYP-substrate interaction based on alignment with bacterial CYP101 (P450cam), the substrate binding residues of which have been identified by X-ray crystallography (Figure 3). Analysis of mutations in experimental studies of numerous CYP isoforms in these SRSs, altogether six sites (SRS1 to SRS6), have been reported to cause altered kinetics for major CYP substrates [18]. As listed in Table 1, residues bonded to the various ligands were the amino acids residing in SRS1, SRS2, SRS4 and SRS5 (Figure 3).

The sequence of the native CYP2C9 with the construct used to determine the 1R9O crystal structure. The N terminus of the wild-type enzyme is depicted on the first line. Modifications are shown in bold type in the truncated 1R9O construct. Helices and strands are labeled and indicated by boxed and underlined text, respectively. Substrate recognition sites (SRSs) are shaded in grey
The relatively more potent binding (lower CDOCKER interaction energy) observed for sulfaphenazole, diacerein, and rhein are likely to be accounted for by two main factors. Firstly, the number of bonds formed in the three ligands was much higher compared to glucosamines and CS4 (9–14 versus 0–4 bonds). A larger number and different types of bonds have resulted in better binding affinity between the ligands and CYP2C9. Interestingly, hydrophobic interactions (pi-pi, amide-pi, and pi-alkyl) appeared to play a role in better binding for sulfaphenazole, diacerein, and rhein, as this was not observed for glucosamines, where interactions were dominated by hydrogen and electrostatic interactions only. Secondly, the three ligands directly interacted with the heme moiety, forming mostly hydrophobic pi-pi interaction (sulfaphenazole), or electrostatic pi-anion interaction (diacerein and rhein). These direct interaction with heme has probably further enhanced stability of interaction, therefore contributing to increased binding affinity observed.
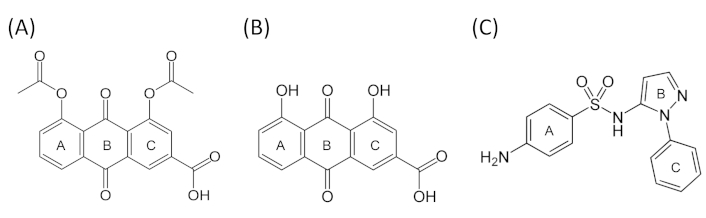
Rhein, an anthracene compound, has an extended conjugated system with two hydroxyl and a carboxyl moieties (bonded to rings A and C) in addition to the central anthracene functionality (Figure 4). It was the most potent inhibitor of CYP2C9-mediated valsartan 4-hydroxylation among the natural anti-arthritic compounds investigated and showed low CDOCKER energy (Table 1). Our docking showed that the compound packed next to helix I and above the heme in a hydrophobic cleft that was formed by VAL113 and PHE114 in the SRS1; ILE205 and LEU208 in SRS2; ASP293, GLY296, and ALA297 in SRS4; and LEU366 in SRS5 (Figure 1A). These residues formed altogether 14 bonds with various structural moieties of rhein. Both VAL113 and PHE114, forming part of the B-C loop, corresponded to ILE113 and PHE114 amino acids of CYP2C2 that have been shown to be critical for substrate binding in site-directed mutagenesis [19–21]. PHE114 formed pi-pi stacking with ring C of anthracene functionality in rhein in our docking model, consistent with similar pi-pi stacks observed for diclofenac in another CYP2C9 homology modeling study [22]. This residue has also been shown by site-directed mutagenesis and physiology-based pharmacokinetic modeling to play an important role in S-warfarin binding and metabolism [23, 24]. Both ILE205 and LEU208 formed pi-alkyl interactions with rhein, and these two residues have been part of hydrophobic side chains along helix F that were involved in interaction with the hydrophobic portion of flurbiprofen in CYP2C9 crystal structure, alongside ASN204 which was involved in direct hydrogen bonding with the substrate [16]. In our model, ILE205 additionally formed a hydrogen bond with O17 of the carboxylate moiety (attached to ring C) of rhein, further contributing to the relatively high affinity binding of this compound to CYP2C9. ASP293, GLY296, and ALA297, located within SRS4 (Figure 3), were all part of the long helix I, which was part of the wall of the heme pocket. ASP293, in particular, was one of the critical residues that formed hydrogen bond with ARG108 in CYP2C9-flurbiprofen crystal structure. Hydrogen bonding of ARG108 with ASP293 and a nearby ASN289 on helix I was demonstrated to stabilize the observed conformation of ARG108 that bonded directly to flurbiprofen in the active site. These interactions position the substrate for regioselective oxidation in a relatively large active site cavity and are likely to account for the high catalytic efficiency exhibited by CYP2C9 for the regioselective oxidation of several anionic non-steroidal anti-inflammatory drugs [16]. Moreover, ASP293 also exhibited hydrogen bonding interactions that were likely to stabilize the adjacent B-C loop formed by the polypeptide backbone at residues 111–114, including VAL113 and PHE114 described above [16]. LEU366, located in the loop region between helix K and β1-4 sheet, resided in SRS5 region (Figure 3) where many residues critical for substrate binding and catalysis have been investigated. For instance, ILE359Leu substitution found in CYP2C9*3 has been shown to cause detrimental effect in substrate catalysis, with reported 3.4- to 26.9-fold reduction in intrinsic clearance for a number of substrates [25]. In addition, LEU366 is also close to LEU363 which interacts directly with diclofenac in CYP2C9-diclofenac homology modeling [22]. It is therefore clear from the discussion above, the high number of bondings and critical locality of the residues involved in direct interaction with rhein, have collectively contributed to the high affinity binding of the ligand in our model.
Similar to rhein, diacerein was shown to locate within the putative active site of CYP2C9 where again a combination of pi-pi stacking and hydrogen bonds orientated the ligand in close proximity to the heme. Docking of diacerein showed hydrogen bonds between the oxygen atom of the two acetyl functions (O22 and O26 respectively) with LEU201 and SER209. Hydrophobic pi-stacking interactions were observed for the aromatic rings of diacerein (rings A, B, and C), including the pi-pi interaction with the hydrophobic PHE114, and the amide-pi bonding with GLY296 and ALA297, along with additional pi-alkyl interactions with the hydrophobic residues VAL113, ILE205, and ALA297. Additional bonding included direct pi-anion interaction with the heme (see Table 1 and Figure 1B). Diacerein was, however, docked in opposite orientation than the docked pose of rhein whereby ring C of the rhein orientated itself toward proximal end of I helix and B-C loop with its carboxyl moiety pointing upward away from the heme (Figure 1A). In the case of diacerein, the ring C was docked closer to I helix distal end, pointing away from B-C loop with its carboxyl moiety pointing downward to the heme (Figure 1B). Nevertheless, both ligands showed the same number of bondings with CYP2C9 and formed the same hydrophobic interaction with VAL113, PHE114, ILE205, GLY296, and ALA297 at the binding site, along with the pi-anion interaction with the heme. Due to this different docking orientation, structural moieties from the two anti-athritic compounds interacting with active site residues were therefore different. As an example, heme interacted with oxygen atom (O17) of carboxylate attached to ring C for diacerein, whereas oxygen atom (O20) of the hydroxyl function of ring A was the interacting atom with the heme in rhein (Figure 1 and Figure 4). From the docked pose of diacerein in our model (Figure 1B), it is evident that LEU201 and SER209, located in F helix and F-G loop respectively and observed only in diacerein but not in rhein, have played crucial role in interacting with the two reactive acetyl moieties of diacerein (via hydrogen bond), and this has probably played a critical part in the observed opposite docking mode of diacerein (to that of rhein) in CYP2C9 cavity. It is therefore apparent that having a unique set of interacting residues (LEU201 and SER209 in diacerein, and LEU208, ASP293 and LEU366 in the case for rhein), together with the opposite orientation of the ligand molecule within the active site, appear to have resulted in different binding affinity and potency as reflected by different CDOCKER interaction energy and IC50 values for these two anti-arthritic compounds.
Sulfaphenazole, in our docking model, demonstrated mainly hydrophobic interaction with a number of active site residues. Benzene ring (ring A) interacted with GLY296, ALA297, and LEU208 via pi-pi, amide-pi, and pi-alkyl bondings respectively. The pyrazole ring (ring B), on the other hand, bonded to LEU208 and LEU366 via pi-alkyl interaction. N-phenyl ring (ring C), which is orientated closer to heme, formed pi-alkyl bonds with VAL113, ALA297, LEU362, and LEU366, and also a direct pi-pi stacking with the heme (Figure 1C and Figure 4). As discussed above, all these residues have been shown to be important in ligand binding and catalysis for CYP2C9 and were also the residues found in the binding of rhein and diacerein described above. These common interacting residues identified in all three ligands indicate their critical role in ligand docking, consistent with the low CDOCKER interaction energy values, as well as the relatively low IC50 values in the in vitro inhibition assay reported in our previous study [13].
As listed in Table 1, both G3S and G6S were involved in the lower number of bondings with CYP2C9 compared to rhein, diacerein and sulfaphenazole (4 versus 9–14). The interacting residues with the two glucosamine molecules were also the residues involved in binding of rhein, diacerein and sulfaphenazole. The residues involved were PHE114, LEU208, SER209, GLY296 and ALA297 (Figure 1D and Figure 1E). The only exception was ARG108, which was hydrogen bonded to G6S in our model. Interestingly, ARG108 has been demonstrated to be involved in charge-charge interaction with carboxylate moiety of flurbiprofen in CYP2C9 crystal structure [16], and shown in site-directed mutagenesis studies to be important in substrate recognition of substrates such as S-warfarin and diclofenac [26, 27]. Despite the importance of ARG108 in ligand binding and catalysis, the lesser number of interacting bonds in both G3S and G6S has likely accounted for the lesser affinity for binding as evidenced by higher CDOCKER interaction energy values and negligible inhibition observed in our previous in vitro inhibition study [13].
Chondroitin sulfate, our last anti-arthritic compound investigated in the docking study, is a natural polymer of disaccharide units with high molecular weight. Docking of this long chain of molecules was not successful in our initial attempts and hence, it was substituted with the smaller disaccharide C4S. Our docking data showed no interaction with the residues at the binding site, even though there was a number of residues located nearby C4S molecule (see 2D diagram in Figure 1F). This was consistent with the high interaction energy (–7.59 kcal/mol) and the experimentally determined IC50 (> 1,000 μM) values.
Based on our data above, we postulate that the inhibition of CYP2C9 by the anti-arthritic compounds investigated would be based on three main interactions. The first one would be the hydrophobic interactions between the phenyl substituents of the compounds with amino acid residues of CYP2C9. The second one would be the hydrogen bonding between donor and acceptor functions found in the ligands (such as alcohol, carboxylate, and acetate functions in rhein and diacerein) and active site residues (such as ILE205, SER 209, and ASP293). Finally, the third one would correspond to ionic (pi-anion) bonding between the negative charge of the alcohol or carboxylate function (as in rhein and diacerein) with the heme. The relative importance of these interactions in the inhibitory effect and the structural factors explaining their selectivity for CYP2C9, however, would merit further investigations. Nevertheless, in view of the prominent roles of these interactions, it is plausible that in developing a new anti-arthritic compound with reduced drug interaction or inhibition liability, designing a compound with lower propensity for hydrophobic, hydrogen, and direct electrostatic bondings (with heme) would represent a promising approach. Among the approaches that can be considered include the replacement of aromatic or pi-containing groups with non-aromatic ring structures, avoiding electron-rich moieties (i.e., amine, thiol, sulfate, etc.) in the hydrocarbon skeleton, and optimizing the nucleophilicity by maneuvering the electron-rich center(s) within the skeletal structure of the developed compound.
It should be noted that this study was performed based on the clues from docking simulations, which was influenced unavoidably by the accuracy of the model used. One of the main factors limiting the accuracy of modeling using rigid protein (as used in the present study) is protein flexibility. There is no proper accounting for conformational flexibility upon ligand binding using this approach. Scoring functions using this method do not consider the contribution of thermodynamic effects on binding free energy, like solvation, long-range interactions, and conformational changes [28]. Docking, therefore, can predict a plausible orientation and conformation of ligands inside the binding site of the receptor, although this method gives only a static picture of receptor-ligand interactions. A deeper insight into the time-dependent properties of protein-ligand complexes could be obtained with the use of MD simulations. There are many examples of successful applications of MD in the characterization of CYP-ligand interactions that have been reported [29, 30].
As a conclusion, we have successfully docked five natural anti-arthritic compounds, diacerein, rhein, glucosamines (G3S and G6S), and C4S to the defined active site of CYP2C9 in the present study. All ligands are bound deep in the active site pocket in close proximity to the heme. Except for chondroitin, all ligands are bonded to residues found in critical secondary structures that form the boundary of the active site cavity as reported in the published crystal structure, including B-C loop, F helix, F-G loop, and I helix. A total of 12 amino acids were involved in the binding and all were critical residues located in four out of six SRSs that have been identified as important substrate binding and catalysis regions in many other CYP isoforms. The better binding efficiency (lower CDOCKER interaction energy) observed for diacerein and rhein compared to glucosamines and C4S are likely due to two main factors: a higher number of bonds between ligand molecule and CYP2C9 active site residues (14 versus 0–4), and direct hydrophobic or ionic interaction with the heme moiety. The binding residues identified in both diacerein and rhein were the residues that also bonded with sulfaphenazole, the specific and potent CYP2C9 inhibitor, confirming their pivotal role in ligand docking. In summary, the in silico data from this study have provided insights into structural features of CYP2C9 critical for inhibition, and formed basis for further exploration of structural determinants for potency and specificity of some commonly used natural anti-arthritic compounds in CYP2C9 inhibition.
C4S: chondroitin disaccharide Δdi-4S
CYP2C9: cytochrome P450 2C9
G3S: glucosamine 3-sulfate
MD: molecular dynamics
OA: osteoarthritis
SRSs: substrate recognition sites
BHT: Data curation, Formal analysis, Investigation, Writing—original draft. NA: Formal analysis, Methodology, Software, Validation, Visualization, Writing—review & editing. YP: Data curation, Formal analysis, Investigation, Writing—review & editing. UDP: Methodology, Data curation, Formal analysis, Investigation, Supervision. CEO: Conceptualization, Data curation, Formal analysis, Funding acquisition, Supervision, Writing—review & editing. All authors read and approved the submitted version.
The authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
The raw data supporting the conclusions of this manuscript will be made available by the authors, without undue reservation, to any qualified researcher.
This work was supported by the Monash University Seed Grant [grant no BCHH-SS-4-02-2010] and the Science Fund [project code 02-02-10-SF0077] of the Malaysian Ministry of Science, Technology and Innovation. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
© The Author(s) 2025.
Open Exploration maintains a neutral stance on jurisdictional claims in published institutional affiliations and maps. All opinions expressed in this article are the personal views of the author(s) and do not represent the stance of the editorial team or the publisher.
Copyright: © The Author(s) 2025. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

View: 1300
Download: 33
Times Cited: 0