Review
Review

Affiliation:
1Engelhardt Institute of Molecular Biology, Russian Academy of Sciences, 119991 Moscow, Russian Federation
2Moscow Institute of Fine Chemical Technology (MITHT), 119571 Moscow, Russian Federation
ORCID: https://orcid.org/0009-0008-9169-1682
Affiliation:
1Engelhardt Institute of Molecular Biology, Russian Academy of Sciences, 119991 Moscow, Russian Federation
ORCID: https://orcid.org/0009-0001-1572-5990
Affiliation:
1Engelhardt Institute of Molecular Biology, Russian Academy of Sciences, 119991 Moscow, Russian Federation
ORCID: https://orcid.org/0000-0002-5399-4219
Affiliation:
1Engelhardt Institute of Molecular Biology, Russian Academy of Sciences, 119991 Moscow, Russian Federation
Email: mdrenichev@mail.ru; room517@eimb.ru
ORCID: https://orcid.org/0000-0001-7624-8801
Explor Drug Sci. 2025;3:1008112 DOl: https://doi.org/10.37349/eds.2025.1008112
Received: January 31, 2025 Accepted: April 22, 2025 Published: May 29, 2025
Academic Editor: Michio Kurosu, University of Tennessee Health Science Center, USA
The article belongs to the special issue Bioactive Molecules from Natural Sources
Pentafuranosylnucleos(t)ides represent a class of natural compounds regulating diverse cell functions being preferably components of biopolymers and also participating as cyclic regulatory low-molecular ligands. Disaccharide nucleosides and related analogs are considered as therapeutically potent compounds for the treatment of cancer, viral diseases, and a variety of metabolic disorders by mimicking a structure of biochemically occurring molecules participating in nicotinamide adenine dinucleotide (NAD+) transformation. Several approaches have been developed on the way to the chemical synthesis of poly(adenosine diphosphate ribose) (PAR), a unique biopolymer taking part in DNA repair and associated functions, that would allow extensive studies of molecular mechanisms of a variety of diseases. The present review consists of the following main parts, the first one including structural characterization, biochemical roles, and chemical synthesis of disaccharide nucleosides from different sources and biopolymers on their basis, the second one describing therapeutic applications of disaccharide nucleosides and their analogs. General conclusion and perspectives are summarized in the last part.
Biochemical modification of nucleosides with additional carbohydrate fragments is widely distributed in nature and imparts them properties of both carbohydrates and nucleosides with their participation in a series of key regulatory properties in cells. Besides commonly known nucleosides and nucleotides, forming DNA and RNA biopolymers, their modified analogs were also observed in the composition of some biopolymers. Being firstly isolated from transfer and ribosomal RNAs (tRNA and rRNA, respectively), modified nucleosides were then observed in messenger and non-coding RNAs [1]. Nucleoside modifications affect many additional structural fragments which are introduced into adenosine (Ado), guanosine (Guo), cytidine (Cyd), and uridine (Urd). One can observe 111 modifications in tRNA, 33 modifications in rRNAs, 17 modifications in messenger RNA (mRNA), and 11 in non-coding RNAs [1]. It is highly possible, that changes in RNA structure are necessary for cell survival and finally viability and development of a whole organism, as well as the progression of various metabolic and cancerous diseases [1, 2]. tRNA possibly represents the most biochemically modified molecule and plays a key role in the translation process, acting as an adaptor molecule to transfer genetic information from mRNA to the amino acid sequence in protein [3–6]. Because of the structural complexity of natural biopolymers, the proper functions of a majority of modified nucleotides are still unknown and will be the object of complex research during the ongoing decades. Poly(adenosine diphosphate ribose) (PAR) can be considered as an important biopolymer, which is involved in various life processes [7–15]. PAR participates in numerous cellular processes such as DNA repair and replication, modulation of chromatin structure, transcription, cell differentiation, and also in the pathogenesis of various diseases such as cancer, diabetes, ischemia, and inflammations. PAR is a homopolymer in which the adenosine diphosphate (ADP) ribose (ADPR) units are linked together by “ribose-ribose bonds”, namely through O-glycosidic bonds between the ribose residues [8, 10, 15]. Short anchor PAR-like fragments of adenosine-5'-diphosphoribosyl groups are covalently attached to a series of regulatory proteins, forming a combined type of biopolymers, nucleoproteins and providing regulatory events by changing protein properties by their PARylation or similar processes [15–18]. Therefore, in the recent review chemical, biochemical and some therapeutic aspects have been described in relation to disaccharide nucleos(t)ides as the objects of striking interest for the study of cell machinery functions and some aspects of their therapeutic applications.
Purine disaccharides 2'-O-β-D-ribofuranosyladenosine-5''-phosphate (Arp, 1a) and 2'-O-β-D-ribofuranosylguanosine-5''-phosphate (Grp, 1b) were isolated for the first time from yeast methionine tRNA (tRNAMet) and several plant sources [8, 10, 19]. This type of nucleoside is located at position 64 of tRNAMet, in its T-loop [8]. The position of the additional phosphate group was determined by periodate oxidation of disaccharide nucleotides with their further β-elimination. It was also shown that an additional ribofuranose moiety is attached to a nucleoside via the β-glycoside bond. The formation and role of Arp in tRNA is still obscure [8]. It is formed by post synthetic transfer of phosphoribosyl moiety from 5-phosphoribosyl-1-pyrophosphate (PRPP) to Ado-64 catalyzed by ribosyl transferase enzyme (Rit). X-ray analysis disclosed the 5-phosphoribosyl moiety occupies a strictly defined place in a small groove on tRNA tertiary surface. The additional phosphate group forms a hydrogen bond with 2-NH2 of neighboring Guo-63. Selective removal of 5-phosphoribosyl moiety led to tRNA beginning to function as an elongator, but less efficiently compared to a native tRNAMet. Thus, it can be proposed that such a modification plays a definite role in the discrimination of initiation-elongation of protein biosynthesis, preventing the formation of tRNA complex with elongation factor EF-1a and GTP [8]. Indeed, we know a little about Arp and Grp activity as low-molecular bioregulators. Therefore, we made some attempts to test their activity on some protein targets (see part about therapeutic application). The first synthesis of Arp and Grp was elaborated by Mikhailov and colleagues [20–23] starting from 3',5'-O-(tetraisopropyldisiloxane-1,3-diyl)ribonucleosides with 1,2,3-tri-O-benzoyl-5-O-phenoxyacetyl-α/β-D-ribofuranose in the presence of tin tetrachloride (SnCl4) in dry dichloroethane (DCE) at 0°С to form disaccharide products in 60–68% yields respectively. Further phenoxyacetyl deblocking with 0.1 М K2CO3 in MeOH and phosphorylation with bis[2-(4-nitrophenyl)ethyl]phosphate and further total deblocking afforded the desired products with 14% and 18% total yields for 1a and 1b respectively (Figure 1).
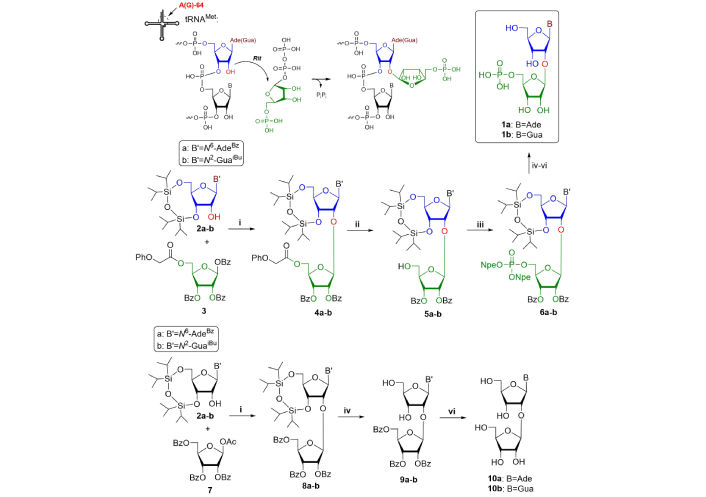
Chemical synthesis and biosynthesis of tRNA disaccharide components. Reagents and conditions: i. SnCl4/DCE, 0°С; ii. 0.1 М K2CO3/MeOH; iii. bis[2-(4-nitrophenyl)ethyl]phosphate/TPSCl/1-Me-1H-Im/Pу; iv. Bu4NF/THF; v. DBU/Pу; vi. NH3/MeOH. tRNA: transfer RNA; tRNAMet: methionine tRNA; Rit: ribosyl transferase enzyme; Bz: benzoyl; SnCl4: tin tetrachloride; DCE: dichloroethane; THF: tetrahydrofuran
The corresponding disaccharide nucleosides (10a-b) can be synthesized in analogous conditions by simple condensation of 3',5'-O-(tetraisopropyldisiloxane-1,3-diyl)ribonucleosides with slight excess of peracylated ribofuranose. The reaction was performed in mild conditions (0°С, DCE, 7–16 h) in 74–82% yield, though at ambient temperature the reaction proceeded faster (30 min) but with lower yields (40–77%) [8].
PAR is a complex branched biopolymer, represented in Figure 2.
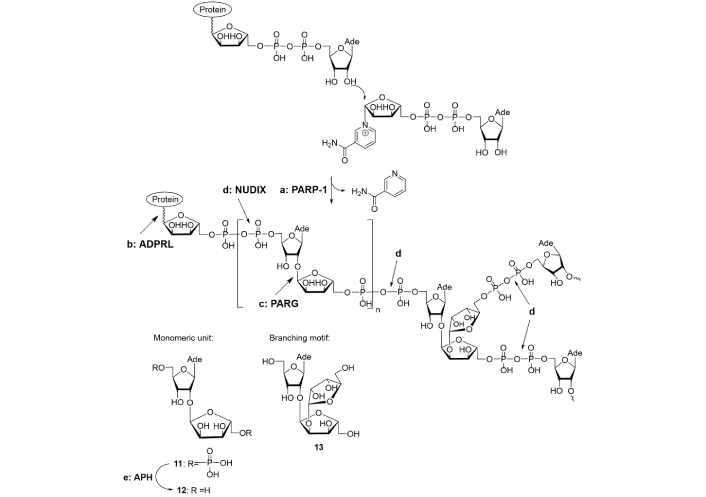
Poly(adenosine diphosphate ribose) (PAR) biosynthesis and depletion enzymes. (a) PARP: PAR polymerase; (b) ADPRL: (ADP-ribosyl) protein lyase; (c) PARG: PAR glycohydrolase; (d) NUDIX: human Nudix hydrolase (hNUDT16); (e) APH: alkaline phosphatase
Note. Adapted with permission from “Poly(ADP-ribose): From chemical synthesis to drug design” by Drenichev MS, Mikhailov SN. Bioorg Med Chem Lett. 2016;26:3395–403 (https://doi.org/10.1016/j.bmcl.2016.06.008). © 2016 Published by Elsevier Ltd.
Native PAR molecules consist of ca. 200 monomeric units, in which linear segments alternate with branching motifs [10]. Enzymatic hydrolysis of the pyrophosphate bonds by snake venom phosphodiesterase (not shown in Figure 2) led to the formation of a mixture of disaccharide and trisaccharide nucleotides, subsequent treatment of which with alkaline phosphatase (Figure 2, pathway e) afforded disaccharide nucleoside (12) and trisaccharide nucleoside (13) respectively [24, 25]. Their structures were determined by NMR spectroscopy. Compound 13 represents the core motif of PAR branching. The common feature of compounds 12 and 13 is the presence of α(1→2) glycosidic bonds (Figure 2). The structure of PAR has some similarity to the structure of nucleic acids, DNA and RNA because antibodies raised against PAR can recognize RNA and DNA [7, 26]. Although PAR exists in an extended conformation in low salt buffers, it may also undergo some structural changes in high salt buffers, showing cooperative hyperchromic helix-coil transitions for a long molecule [26]. Three-dimensional structural information available by investigations of isotopically labeled PAR with nuclear Overhauser enhancement spectroscopy (NOESY) has revealed adenine base location in anti-conformation and thus heterocyclic moieties in PAR are accessible both for inter- or intramolecular interactions, that is in agreement with possibilities of helix-coil transitions, established by CD-spectroscopy [27]. It is not therefore surprising that PAR functions in cooperation with DNA and RNA structures and respectively associated proteins [7]. Thus, PAR interacts directly or indirectly with a series of regulatory and repair proteins and enzymes and modulates their functions [7, 28–31]. Covalent modification of proteins proceeds either via the formation of O-glycoside bond (Glu/Asp, Ser/Thr residues) or via the formation of N-glycoside bond (Lys/Arg, Asn/Gln residues) with short ADPR moiety being an anchor for PAR building up [32]. Covalent modifications of serine, acetyllysine, phosphoserine and covalent modification of DNA and RNA at guanine base were also described [33]. On molecular level functional role of PAR is based preferably on ionic interactions with target proteins and binding with nucleoside parts of ADPR-ADPR junction or terminal ADPR fragment. Charged lysine- and arginine-rich motifs are essential for ionic interactions of histones with PAR and thereby afford injured DNA unwinding for repair [34].
Biosynthesis and cleavage of PAR is catalyzed by several enzymes of various classes (Figure 2). In cellular nuclei, PAR is synthesized by PAR polymerases (PARPs) recruiting nicotinamide adenine dinucleotide (NAD+) as a substrate (Figure 2, pathway a). Nowadays, PARP family numbers about 17 enzymes. PARP-1 (EC 2.4.2.30) is responsible for the synthesis of 90% of PAR in cells and can be considered as the most widespread PARylating enzyme in higher eukaryotes. The first step associated with PAR biosynthesis is a covalent attachment of the first ADPR moiety to an acceptor amino acid. Further biosynthesis of PAR consists of elongation of PAR chain by joining of additional ADPR monomers; and generation of branching points [7, 35–39]. In fact, enzymes catalyzing these two steps strongly differ in structure and function. PARP-1 cannot operate with small threads of PAR, while the other representatives of the PARP family can transfer only short ADPR fragments. This leads to enormous complexity of PARP functions in cells. PAR is cleaved by three enzymes, viz., (ADP-ribosyl) protein lyase (ADPRL; Figure 2, pathway b), PAR glycohydrolase (PARG; Figure 2, pathway c), and NUDIX hydrolase, cleaving pyrophosphate linkages [38, 40–43]. O-Glycosidic bond between adenosine and ribofuranose moieties in PAR-polymer is split by PARG. ADPRL catalyzes the cleavage of O-glycosidic bond between ADPR residue and proteins [38]. Thus, at least 20 enzymes are involved in the biosynthesis and degradation of PAR polymer.
In general, PAR can interact with more than 500 proteins bearing specific binding sites [11, 12, 15, 30, 31]. Its biological role is well understood but the precise mechanism of action and interaction with target proteins are still unclear. Therefore, enzymatic or chemical synthesis of well-defined linear PAR oligomers is needed for more tight understanding of its interactions with proteins. Current enzymatic methods for the preparation of PAR oligomers are not reliable and afford a mixture of compounds with different molecular weights and even branching points. PAR fragments of sufficient homogeneity could be obtained only by multiple chromatographic purification steps [44, 45]. Therefore, the development of the synthesis of regular PAR oligomers is a challenging problem for chemists. The complexity of this problem is associated with several hurdles: generation of α(1→2) glycosidic bond between adenosine sugar moiety and additional ribofuranose fragment; and their joining with labile pyrophosphate bonds. The first obvious step in this way could be the chemical synthesis of disaccharide nucleosides and orthogonally protected blocks on their basis by the formation of an O-glycosidic bond between a nucleoside and monosaccharide and coupling of a protected disaccharide with a heterocyclic base analogously to a procedure earlier proposed by Vorbrüggen and Ruh-Pohlenz [46]. The presence of a neighboring 2-O-acyl group leads to the formation of intermediate 1,2-acyloxonim ion which determines the stereochemistry of this reaction and yields β-glycosidic bond formation in the case of glycosylation of both nucleoside and heterocyclic base [8]. Using this strategy, 2'-O-α-D-ribofuranosyladenosine, a component of PAR polymer, was obtained for the first time by Mikhailov et al. [47]. 1,1,3,3-Tetraisopropyldisiloxane-1,3-diyl (TIPDS)-protected N6-benzoyladenosine (3a) reacted with peracylated arabinofuranose (14) to form α(1→2)-glycoside bond under SnCl4 catalysis, as depicted in Figure 3.
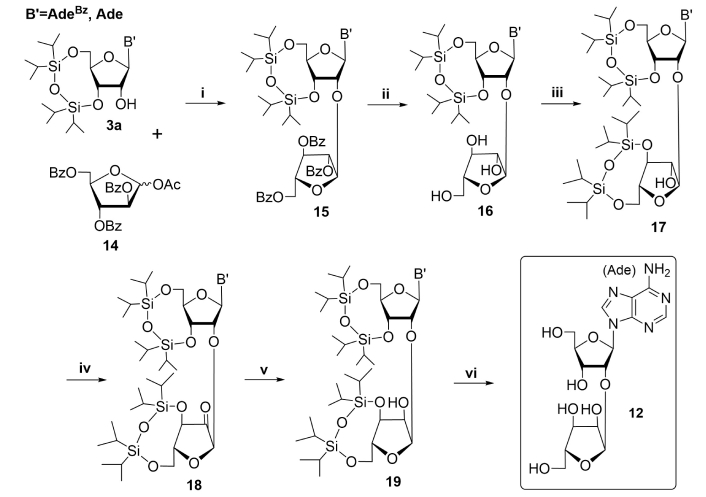
Chemical synthesis of PAR monomeric unit. Reagents and conditions: i. B' = AdeBz: SnCl4/DCE, nitrogen atmosphere, 0ºC, 24 h, 62%; B' = Ade: SnCl4/DCE, nitrogen atmosphere, 0ºC, 3 h, 64%; ii. 0.1 M MeONa/MeOH, 10ºC, 40 min, 47% (B' = AdeBz), or 8 M MeNH2/EtOH, 20ºС, 24 h, 87% (B' = Ade); 77% (B' = AdeBz→B' = Ade); iii. TIPDSCl2/pyridine, 35ºС, 20 h, 69% (B' = AdeBz), 80% (B' = Ade); iv. DMSO/Ac2O, 20ºС, 24 h; v. NaBH4/EtOH, 0ºC, 1 h, 69% (B' = AdeBz) for two steps, 62% (B' = Ade) for two steps; vi. NH3/MeOH, 20ºС, 2 days (B' = AdeBz), Bu4NF∙3H2O/THF, 20ºС, 1 h, 93% (B' = AdeBz) or Et3N·3HF, Et3N/THF, 20ºС, 24 h, 78% (B' = Ade). PAR: poly(adenosine diphosphate ribose); SnCl4: tin tetrachloride; DCE: dichloroethane; TIPDS: 1,1,3,3-tetraisopropyldisiloxane-1,3-diyl; THF: tetrahydrofuran; Bz: benzoyl
Note. Adapted with permission from “Poly(ADP-ribose): From chemical synthesis to drug design” by Drenichev MS, Mikhailov SN. Bioorg Med Chem Lett. 2016;26:3395–403 (https://doi.org/10.1016/j.bmcl.2016.06.008). © 2016 Published by Elsevier Ltd.
This made possible an opportunity to reverse group configuration locating around 2''C-atom of additional arabinofuranose moiety by at least a four-step procedure to convert it to ribofuranose one with group configuration at O-glycosidic bond being not affected. First step, sodium methoxide afforded partial debenzoylation with the formation of N6-benzoyl derivative (16, B' = AdeBz) in medium 47% yield due to the partial desilylation. The use of milder debenzoylation conditions (4 M or 8 M methylamine in ethanol, ambient temperature, 24 h) afforded compound 16 (B' = Ade) nearly twice as high yield and thus allowed avoiding undesirable side reactions [48, 49]. The second step was selective protection of 3'- and 5'-hydroxyl moieties of arabinose by TIPDS protection to afford compound 17. The key steps of configurational inversion at C-2 of the extra arabinose residue in compound 17, were successively conducted by oxidation of the 2''-hydroxyl group to a ketone (18) and its diastereoselective reduction with sodium borohydride [47, 49]. In the final step, silyl protective groups were removed by the treatment with tetrabutylammonium fluoride with high yields of PAR-disaccharide unit. The use of partially blocked nucleoside (Figure 3, B' = Ade, route i), benzoyl groups removal with methylamine in ethanol (Figure 3, route ii), and silyl groups removal with triethylamine trihydrofluoride (Figure 3, route vi) increased the overall yield of the resulting product more than 1.5 times [49].
Another synthetic challenge, the production of the branching motif of PAR required an elaboration of non-standard approach, which was first described by Kistemaker et al. [35]. An introduction of the third ribofuranose moiety into disaccharide structure appeared to be complicated by the steric hindrance of 2''-hydroxyl group in protected 2'-O-α-D-ribofuranosyladenosine (19) [47–49]. 2''-O-α-D-Ribofuranosyl-2'-O-α-D-ribofuranosyladenosine thus was synthesized recently by a different route, starting with trisaccharide glycosyl donor (20) (Figure 4) [35].
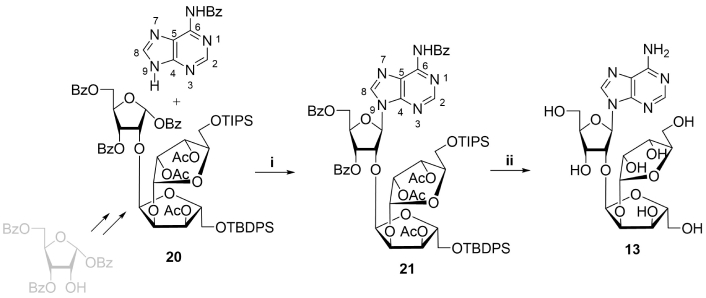
Chemical synthesis of PAR branching motif. Reagents and conditions: i. AdeBz, BSA, HClO4-SiO2, MeCN, reflux, 4 h, 63%; ii. a) HF, pyridine, r.t., 1 h, b) TBAF/THF, r.t., 16 h, c) 30% NH4OH, 50°C, 16 h, 57% on three steps. PAR: poly(adenosine diphosphate ribose); TBAF: tetrabutylammonium fluoride; THF: tetrahydrofuran; Bz: benzoyl; BSA: N,O-bis(trimethylsilyl)acetamide
Note. Adapted with permission from “Poly(ADP-ribose): From chemical synthesis to drug design” by Drenichev MS, Mikhailov SN. Bioorg Med Chem Lett. 2016;26:3395–403 (https://doi.org/10.1016/j.bmcl.2016.06.008). © 2016 Published by Elsevier Ltd.
A key step of the synthesis was glycosylation in the presence of chloric acid of N6-benzoyladenine with selectively protected trisaccharide donor (20), prepared by a multistep procedure. Further removal of protective groups in resulting 21 afforded trisaccharide nucleoside (13) in 57% yield.
A possible general strategy for the chemical synthesis of PAR oligomers may be based on a successive coupling of divergently blocked disaccharide synthons, containing phosphorous residues. Because of two primary and three secondary groups in disaccharide nucleosides, a multistep approach of orthogonal protection of free nucleoside hydroxyls is required. In this way, alternative synthetic approaches to form α(1→2) glycosidic bonds are highly desirable to reduce the quantity of steps in synthetic procedures. The reaction of benzyl or allyl protected ribofuranose derivatives with partially silylated adenosine proceeded with high stereoselectivity to form α(1→2) glycosidic bond in good yields under various conditions [50, 51]. Unfortunately, the combination of these two approaches with pyrophoshorylation method was not fruitful in producing PAR oligomers.
An alternative procedure originally proposed by Dmitri V. Filippov research group [52] included the synthesis of three orthogonally protected phosphorylated blocks with their further regioselective condensation to form short PAR oligomers (n = 0–2) (Figure 5). PAR monomeric unit was generated from 5'-O-phosphoramidite-5''-O-phosphate of orthogonally protected ribofuranosyladenosine synthon (31) which was obtained by multistep procedure starting from α(1→2) ribofuranosylribofuranose (25), which was then condensed with N6-benzoyladenine to form disaccharide nucleoside scaffold. The building block 31 was obtained on at the preparative scale (4 mM) by multistep procedure, involving regioselective glycosylation step, and then was used in solid-phase synthesis of PAR oligomers similar to a standard oligonucleotide protocol with β-cyanoethyl group removal by DBU [52].
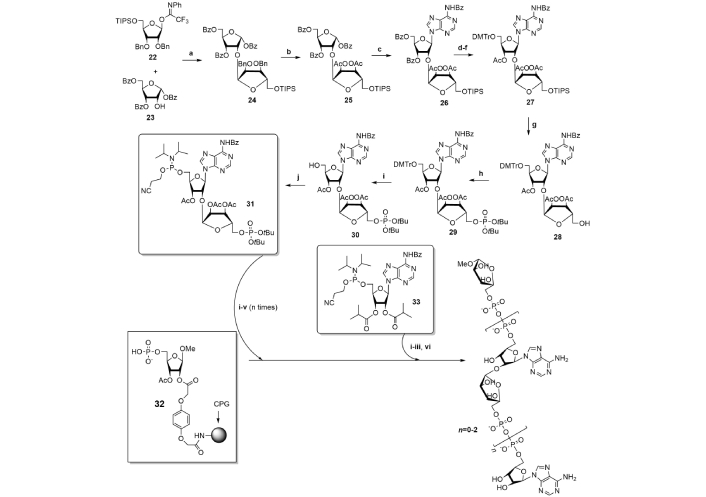
Synthesis of regular PAR oligomers. Reagents and conditions: a. TMSOTf/DCM, –78°C, 86%; b. H2, Pd/C, tBuOH/dioxane/H2O then Ac2O/Py, 85%; c. HClO4-SiO2, MeCN/BSTFA, N6-Ade, reflux, 82%; d. Py/EtOH/NaOH (3:2:3, v/v/v); e. DMTrCl/Py; f. Ac2O/Py; d–f. 83%; g. Et3N·3HF/Et3N, Py, 75%; h. (tBuO)2PNiPr2, 1-Me-Im·HCl, 1-Me-Im, DMF, then tBuOOH, 79%; i. TFA/DCM, 91%; j. 2-cyanoethyl-N,N-diisopropylchlorophosphoramidite, DIPEA/DCM, 71%. PAR-synthesis: i. ETT/MeCN; ii. CSO/MeCN; iii. DBU/DMF; iv. HCl/HFIP; v. pyridine/MeCN; vi. NH4OH. PAR: poly(adenosine diphosphate ribose); ETT: ethylthiotetrazole; CSO: (1S)-(+)-(10-camphorsulfonyl)-oxaziridine; HFIP: hexafluoroisopropanol; CPG: controlled pore glass; Bn: benzyl; BSTFA: N,O-bis(trimethylsilyl)trifluoroacetamide; DMTrCl: 4,4'-dimethoxytrityl chloride; Bz: benzoyl; DIPEA: N,N-diisopropyldiethylamine; DMF: N,N-dimethylformamide; TMSOTf: trimethylsilyl trifluoromethanesulfonate
Note. Adapted with permission from “Poly(ADP-ribose): From chemical synthesis to drug design” by Drenichev MS, Mikhailov SN. Bioorg Med Chem Lett. 2016;26:3395–403 (https://doi.org/10.1016/j.bmcl.2016.06.008). © 2016 Published by Elsevier Ltd.
Hydroquinone-O,O'-diacetic acid was selected as the optimal linker for immobilization due to its resistance to cleavage by DBU, which was used in the step of removal of β-cyanoethyl group. ADPR dimer and trimer were isolated with 35% and 29% yields respectively and thus the first synthesis of regular PAR oligomers was successfully approved. It would not be wrong to assume, that elaboration of every synthetic procedure demanded non-standard thinking, inspiration and a great deal of endurance, accuracy and potency as the literature data show, what a difficult problem was to synthesize even short PAR oligomers. Therefore, the synthesis of long regular PAR oligomers is still an insistent task for modern synthetic chemistry.
Intramolecular cyclization is another bioregulatory role of NAD+ which affords a series of cyclic ADPR (cADPR), the general function of which being immune regulation that is tightly associated with cytoplasmic Ca2+ release and entry [53–58]. Toll/interleukin-1 receptor (TIR) is a family of ancient immune modules present in all domains of life and possessing a NAD+ cyclization catalytic activity (Figure 6).
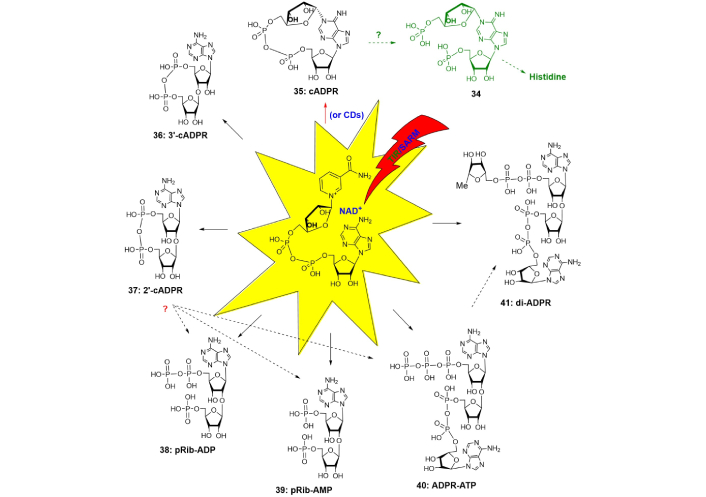
Cyclic NAD+-metabolite. Dotted arrow indicates possible metabolic pathways, which consist of more than one biosynthetic step (indicated by solid arrow). cADPR: cyclic adenosine diphosphate ribose; TIR: Toll/interleukin-1 receptor; NAD+: nicotinamide adenine dinucleotide; pRib: 5-phosphoribose
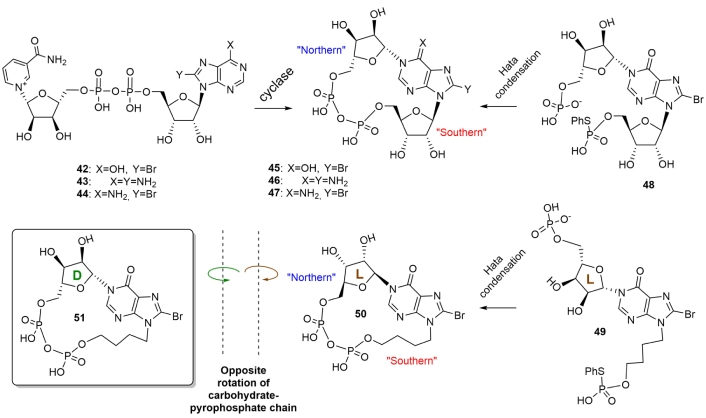
SARM1 belongs to the most evolutionally conserved type of TIR-receptor, expressed also in human cells. TIR/SARM undertake NAD(P)+ degradation and cyclization [58–63]. At a glance, on molecular level, NAD+-cyclization is hypothesized to take part in the development of innate immunity by affection on cytoplasmic Ca2+ release and entry. 1N-Ribofuranosyl-ADP or cADPR (35), the most widespread regulator, is a cyclic dinucleotide that is produced from NAD+ by intermolecular transfer of ribosyl moiety from nicotinamide of NAD+ to position 1 of adenine. In humans and mammalians, this transfer is catalyzed not by TIR, but by ADP-ribosyl cyclases CD38 and CD157 (Figure 6, red arrow) [56, 64, 65]. Firstly, discovered in sea urchins, cADPR then appeared to participate in wider biological roles, being a universal Ca2+ mobilizing second messenger, especially receptor stimulation in immune cells. Inside T-cell cADPR is involved in sustained Ca2+ release and also in Ca2+ entry. It mobilizes intracellular Ca2+ that in turn controls a diverse range of highly regulated cellular processes [64]. The physiological receptor for cADPR is the ryanodine receptor (RyR), while Ca2+ entry seems to be quite intricate and includes a series of receptor proteins with various functions [64]. Chemical lability of N1-linkage of cADPR to hydrolysis can introduce distortions into experimental data interpretation and make its chemical synthesis a complicated problem. A replacement of cADPR with its stable analogs can partially unravel mechanistic aspects of cADPR interaction with receptors, study cADPR/Ca2+ signaling pathways, and develop new medications. An increased basicity of 1N-substituted purine can even provide cADPR molecule to self-catalyze its own depletion [66]. Changing of purine ring properties can be applied to strengthen N1-linkage of cADPR analogs. Therefore, this can be achieved by the introduction of specific electronoacceptor substituents into the purine ring in the surroundings of 1N-gycosidic bond. An alternate synthetic protocol which can be considered is a replacement of easily eliminating ribose moiety with an alkyl analog to generate a more chemically stable 1N-alkyl bond. NAD+-cyclizing enzymes from molluscs were non-specific to chemically modified NAD+(NADP+)-analogs and utilized them to afford plenty of cADPR analogs (Figure 7). The nature of substituents both in purine or carbohydrate rings appeared to affect agonistic and antagonistic effects [56, 64].
8-Bromo-N1-cycloinosinediphosphoribose (8-Br-N1-cIDPR, 45) evoked Ca2+ signaling in the human T-lymphoma cell line Jurkat and in primary rat T-lymphocytes [64]. Ca2+ signaling induced by 8-Br-N1-cIDPR, consisted of Ca2+ release and Ca2+ entry, while 8-Br-cADPR (8-bromo-cADPR) manifested antagonistic effects on Ca2+ release. A replacement of N (northern)-ribose with an acyclic fragment or addition of phosphate groups at S (southern)-ribose fragment dramatically decreased the receptor stimulating activity of cADPR analogs [56, 64]. The use of 2'-deoxy-cADPR analogs (modification at S-ribose) strengthened their activity on sea urchin receptors but not in human T-cells [56]. In general, the structure-activity relationship of cADPR-induced Ca2+ release revealed by cADPR analogs is still evolving and presents a complex picture depending on the structure of compounds and the biological origin of receptors. Chemical synthesis of cADPR analogs is associated with lower regioselectivity compared to the enzymatic method due to the possibility of the formation of several conformers. These conformers can differently bind to receptors. It was shown by computational studies [65] the possibility of the two conformers for cADPR with various torsion angles χ O4'-C1'-N9-C4 in “southern” and “northern” ribose moiety (χS = 46.8°, χN = 9.4° for natural conformer, χS = –6.2°, χN = 57.6° for unnatural conformer, the two ones being syn/syn). Fortunately, the energetic barrier between the two conformers was large enough to presumably form natural conformation. L-Ribose, the mirror image of D-ribose, at the N1-position should exhibit the opposite lowest energy conformation to its D-ribose counterpart [65]. Therefore, according to NMR, only one conformer was formed during intramolecular cyclization of 1N-(5-O-phospho-D-ribofuranosyl)-AMP under Hata reaction conditions (nucleoside 5'-phosphate-5-thiophosphate-I2/pyridine/molecular sieves) (Figure 7). Stereoisomerism of 1N-ribofuranose can determine specific rotation of molecular counterparts, which was demonstrated by the replacement of S-ribose with acyclic fragment, giving more conformational freedom to the obtained molecules [65]. The hydrogen bond between the N-ribose 2''-hydroxyl group and the hypoxanthine 6-oxo group managed conformer formation. By replacement of the N-D-ribose with L-ribose sugar, the cyclization of the linear precursor gave a cyclized product with opposite specific rotation and different biological activity [66]. “L-cIDPR” (50) was a less potent inhibitor of CD38-mediated cADPR hydrolysis than D-isomer and could be hydrolyzed by high concentrations of CD38. 8-Br-L-cIDPR was a potent inhibitor of CD38 acting in micromolar concentrations but was not utilized by the enzyme suggesting a non-competitive mechanism of inhibition. A range of disaccharide nucleosides and nucleotide regulators of immune response in plants have been synthesized at various times, with the most tedious one proposed for disaccharide 5',5''-dinucleotide (34). The multistep procedure consists of 15 steps of successive protection/deprotection steps with 5-O-phosphorylation near the end of the synthetic rote [67, 68]. A proposed protocol for the synthesis of 36 and 37 may include intermolecular phosphate condensation under Hata conditions. Structurally related compounds pRib-AMP/ADP (2'-(5''-phosphoribosyl)-5'-adenosine mono-/di-phosphate) (38–41), which trigger immune signaling in plants [67, 68] are metabolic precursors of various disaccharide nucleosides alternatively to NAD+ and may be formed from 2'/3'-cADPRs by depletion of the later. N1-Phosphoribosyl-AMP (34) (Figure 6), structurally similar to cADPR, is formed by an unusual reaction that joins ATP to PRPP to afford metabolic precursors in the biosynthetic pathway of histidine in plants [3]. Its structural similarity to cADPR, therefore, may indicate a relationship between various pathways of amino acid biosynthesis and cADPR utilization.
There are convincing medicinal proofs, that PAR is a participant in the pathogenesis of a range of diseases, i.e. cancer, diabetes, ischemia, and inflammations [69–75]. PAR-metabolizing enzymes are potential levers of PAR biosynthesis, with two major enzymes being PARP-1 and PARG. PARP-1 is the most studied enzyme among the PARP family. Being a key protein regulating base excision repair (BER) and DNA single-strand breaks (SSB) [39], PARP-1 can be considered as the prospective enzymatic target for the design of new anticancer and anti-inflammatory drugs [76–80]. It represents a six-domain construction: automodification domain (AMD) with 22 kilodaltons, three DNA-binding domains (Zn1, Zn2, and Zn3) with 42 kilodaltons total weight and catalytic subunit with a molecular mass of 54 kilodaltons (cat-PARP) [39, 80]. The use of alkylating drugs in the chemotherapy of tumors injures DNA both in healthy and cancer cells with further DNA repair in both cases. The use of PARP-1 inhibitors in combination with alkylating agents is expected to decrease the concentration of the latter and, hence, decrease the toxicity of chemotherapy. Breast cancer-associated genes BRCA1 and BRCA2 play an important role in the repair of double-strand breaks (DSB) and suppress tumor growth [11, 39, 69, 79]. The loss of one gene function (PARP-1 or BRCA1/2) is not a dramatic event, but the loss of both genes (PARP-1 and BRCA1/2) is lethal for a cell. In BRCA1/2 deficient cells, PARP-1 is upregulated and therefore in these cases, PARP-1 inhibitors can be used as single agents for the treatment of BRCA-deficient tumors [11, 39, 69, 79]. Among the PARP family PARP-2 comprises 60% similarity with PARP-1 in amino acid composition, being the most structurally related to PARP-1. Despite PARP-1 and PARP-2 differ by domain composition, relating to interactions with proteins, a structural similarity of catalytic centers can provide overlapping of their functions and their partial compensation in the case of arrest of one of them, which was evidenced by experimental procedures [81, 82]. For example, a simultaneous elimination of genes encoding PARP-1 and PARP-2 leads to the death of an organism in the embryonal stage [83]. The use of PARP inhibitors can strengthen the action of DNA-injuring drugs on cancerous cells and mitigate treatment toxicity due to the decrease in the concentration of chemotherapeutic compounds, used in combination with PARP-1/PARP-2 inhibitors [84]. Natural nucleosides and nucleotides manifest low PARP-1 inhibition activity, but disaccharide nucleosides seem to be an advantageous class of PARP-1 inhibitors due to their cell penetrability and low cytotoxicity [85]. A possible molecular mechanism of their action can be associated with mimicking of PAR disaccharide core structure. Therefore, a series of the structurally related 2'-O-α-D-arabinofuranosyl- and ribofuranosyladenosine analogs was obtained by various research groups as inhibitors of PARP-1 enzyme (Table 1) [85, 86].
Inhibition of human recombinant poly(adenosine diphosphate ribose) polymerase (PARP)-1 by some synthetic nucleoside analogs with different carbohydrate moiety
| Carbohydrate moiety | Compound | Base | Modification | IC50 µM |
|---|---|---|---|---|
 | 52 | AdeMe | 2'-O-α-D-Ara (53) | 93.4 ± 3.7 |
| 53 | AdeMe | > 250 | ||
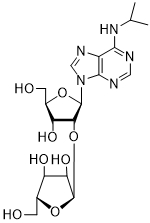 | 54 | AdeiPr | 2'-O-α-D-Ara (55) | 158.2 ± 17.7 |
| 55 | AdeiPr | > 250 | ||
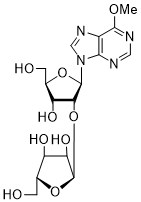 | 56 | PurOMe | 2'-O-α-D-Ara | > 250 |
| 57 | PurOMe | |||
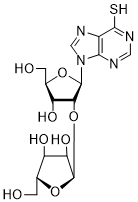 | 58 | PurSH | 2'-O-α-D-Ara | > 250 |
| 59 | PurSH | |||
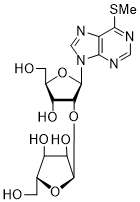 | 60 | PurSMe | 2'-O-α-D-Ara | > 250 |
| 61 | PurSMe | |||
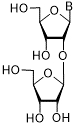 | 62 | Thy | 178 ± 6 | |
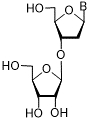 | 63a | Ura | 5-F | 45 ± 3 |
| 63b | Ura | 5-I | 38 ± 4 | |
| 63c | Thy | 5'-PO3H | > 2,000 | |
| 63d | Thy | 5''-PO3H | 216 ± 56 | |
| 3'-Rib-α-Thd | 64 | Thy | > 2,000 | |
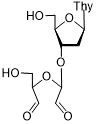 | 65 | Thy | 25 ± 3 | |
| 3-Amino benzamide | 57 ± 8 |
Thd: thymidine; AdeMe: N6-methyladenine; AdeiPr: N6-isopropyladenine; PurOMe: 6-methoxypurine; PurSH: 6-thiopurine; PurSMe: 6-methylthiopurine; Thy: thymine; Ura: uracil; Ara: arabinofuranose; IC50: half maximal concentration, at which 50% enzymatic activity reduction is observed
Several PARP-1 inhibitors active in the micromolar range of concentrations were found among purine and pyrimidine disaccharide nucleosides. The hit-compounds were the most structurally related to the PAR monomeric unit and contained an additional methyl or isopropyl moiety at position N6 of purine. They manifested half maximal concentration (IC50) values at 93–158 µM (see compounds 52, 54) [86].
The introduction of arabinofuranosyl moiety (see compounds 53, 55) drastically lowered PARP-1 inhibition [86]. The replacement of purine residue with pyrimidine one and modifications in the structure of disaccharide scaffold increased PARP-1 inhibition several times. Compounds bearing ribofuranose moiety at 2'-O-hydroxyl group manifested similar activity with purine derivatives, while 3'-O-β-D-ribofuranosyl deoxythymidine appeared to be several times more active [85]. The addition of phosphate groups reduced the inhibitory effect. The introduction of a bulky iodine substituent into position 5 of 3'-O-β-D-ribofuranosyl deoxynucleoside instead of methyl group had the most pronounced effect on PARP-1 inhibition. At the same time, PARP-1 appeared to be sensitive to N-glycoside bond configuration as α-thymidine derivative with drastically lowered activity. The most intriguing feature of pyrimidine disaccharide nucleoside PARP-1 inhibitors was their ability to accumulate in cell nuclei and inhibit PARP-1 inside them, as was shown by our experiments on SKOV-3 cancerous cell line [85]. Low toxicity of disaccharide compounds is probably explained by an absence of enzymes hydrolyzing O-glycosidic bonds inside cancer cells. It was also shown that the dialdehyde derivative of 3'-O-β-D-ribofuranosyldeoxythymidine manifested high PARP-1 inhibition potency but was rather toxic leading to cell agglutination (PARP-2 inhibition of the obtained compounds is now under study) [87].
A molecular mechanism of pyrimidine nucleoside action can be associated with hydrogen bond formation of pyrimidine carboxamide moiety with backbonds of serine (Ser-904) and glycine (Gly-863) in PARP-1 active site. This type of molecular interaction is similar to a variety of PARP-1 inhibitors used in medicinal practice and bearing the carboxamide pharmacophore group. The most important structural motifs in PARP-1 inhibitors are carboxamide fragments and aromatic moieties [77, 86–88]. Therefore, the structure of heterocyclic base is crucial for PARP-1 inhibition as a natural nucleoside thymidine (Thd) possesses PARP-1 inhibition activity. PARP-2 inhibition activity was recently described for some 5-substituted uracil analogs [84]. According to molecular docking data, their mechanism of action is highly possible due to hydrogen bond formation between pyrimidine carboxamide moiety and backbonds of serine (Ser-470) and glycine (Gly-429) in PARP-2 catalytic site.
It was previously shown in a series of investigations, that depending on its length and weight PAR can associate with various protein targets [15, 16, 28]. PARP-1 overactivation during exposure of cells to a toxic stimulus produces toxic levels of PAR [89]. Once being located in cytosol, PAR molecule triggers cell death by parthanatos mechanism (derived from the Greek Θάνατος, “death”). It is a programmed cell death, which is distinct from such processes as necrosis and apoptosis. Parthanatos is involved in the progression of a range of metabolic disorders such as Alzheimer’s/Parkinson’s disease, stroke, heart attack, and diabetes. As the name implies, PAR is the most electronegative natural polymer (in pyrophosphate pK1 < 2.0, pK2 = 2.64) [90] with two negative charges per monomeric unit. Other biopolymers, such as DNA and RNA, contain only one phosphate group per monomeric unit and have only one negative charge at neutral pH. PAR structure has much in common with natural polysaccharides heparin and heparan sulfate as the presence of sulfate and carboxyl groups defines the substantial negative charge of these biopolymers under neutral conditions. Heparin is the mostly charged polysaccharide with –2.7 on the disaccharide unit (–1.35 on the monosaccharide unit). Therefore, charged biopolymers may mimic ionic interactions with PAR-binding proteins, thus having the potency to affect biochemical processes associated with PAR and modulate some cellular functions (Figure 8).
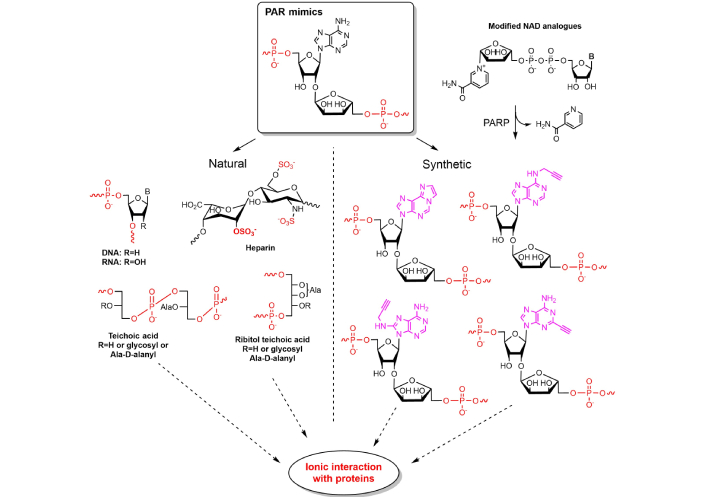
Natural and synthetic biopolymers mimicking PAR. Dotted arrows indicate multiple interactions with various protein targets, solid arrow indicates classification of origin of PAR mimics. PAR: poly(adenosine diphosphate ribose); Ala: alanin; NAD: nicotinamide adenine dinucleotide; PARP: PAR polymerase
Inhibition of PARG may be essential for the study of PAR dynamics and metabolism. The known PARG inhibitors can be both of monomeric or polymeric nature, such as ADPR-analogs ADP-(hydroxymethyl)pyrrolidinediol (ADP-HPD) and poly(etheno-ADPR) respectively [37, 91]. PARP-1 also manifests wide substrate specificity to the nicotinamide moiety of NAD+. Thus, modified NAD+ analogs may be utilized by the enzyme as efficiently as NAD+. Substitution of nicotinamide moiety with compatible aromatic or structurally related groups can be used for colorimetric detection of PARPs and other NAD+ consuming polymerases [92]. Chain terminators, which are NAD+ analogs lacking one or both hydroxyl groups at ribose of adenosine moiety in NAD+, are biochemically utilized by PARP-1 and provide an additional chemical tool for protein labeling and study of PAR’s mechanisms of action [93]. It was found, that 2-hydroxyl group of adenosine moiety is essential for PAR formation. Nevertheless, NAD+ derivatives without 2'- or 3'-hydroxyl group at adenosine moiety can be incorporated into PAR in the presence of NAD+ [93]. Therefore, chemically modified NAD+ can be used as substrates for enzymatic synthesis of chemically modified biopolymers, mimicking PAR functions.
All the described data suggest that ADPR and NAD+ may be a part of a delicate biochemical mechanism regulating transcription and translation in cells. ADP-ribosylation in host-virus conflicts triggers various routes of cellular resistance to infection. PARP-10-mediated modification of RNA ends could have an immune function as its expression inhibits Venezuelan equine encephalitis virus (VEEV) translation and participates in modulating inflammation. It was shown that VEEV and SARS-CoV macrodomains encoded within the non-structural proteins can remove ADP-ribosylation from PARP-10-modified RNA, counteracting PARP-10 activity [94]. It is hypothesized that the PARP-10 RNA recognition motif (RRM) specifically binds and ADP-ribosylates viral RNA, distinguishing it from a host RNA. It was shown in recent studies that SARS-CoV Mac1 reverses the PARP-14 (ART14)-catalyzed transfer of ADPR on signaling proteins thus abolishing immune response. Mac1 is a domain of a larger viral protein known as nonstructural protein (NSP3) and belongs to the family of macrodomains, involved in the post-translational modification (PTM) ADPR. Thus, ADPR represents the simplest disaccharide form of PAR and a platform for the design and synthesis of antivirals. Highly efficient disaccharide inhibitors of the coronaviral Mac1 domain have been synthesized for the first time by Dmitri V. Filippov research group using the phosphate-phosphoramidite coupling strategy (Figure 9) [95].
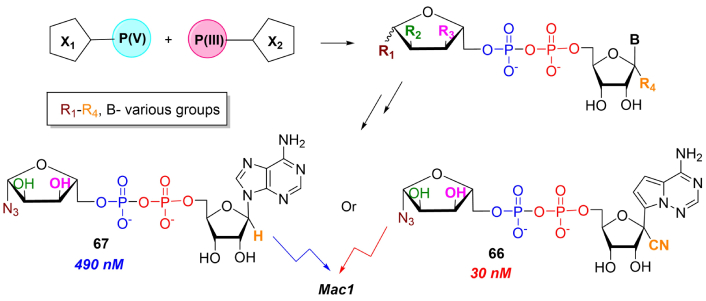
Synthesis of ADPR analogs as potential antiviral compounds. Black arrows indicate synthetic steps. ADPR: adenosine diphosphate ribose
Using this synthetic approach, the nanomolar-range efficient compounds were obtained by combination of two pharmacophore groups based on remdesivir 5'-monophosphate (see compound 66) and modified carbohydrate 5'-phosphate. The proposed combinatorial approach allows the synthesis of libraries of novel compounds and therefore may have applications in antiviral therapy due to its potency to overcome drug resistance associated with mutations in the viral genome. Alternate ADPR synthetic modifications with propargyl group can be further functionalized by biotin and fluorescent dye and possibly applied in isolation and detection of viral and other ADPR-interacting proteins [51].
An enormous deal of bioregulatory events in cells are associated with pentafuranosylnucleosides which are mostly components of biopolymers. These are formed by pentafuranose transfer on a variety of glycosyl-acceptors including tRNAs, NAD+, and other nucleos(t)ides and proteins. Arp (1a) and Grp (1b) were found in yeast tRNA and probably participate in the formation of tRNA spatial structure. Despite their functions in tRNA being audibly studied, the biological role of Arp and Grp as low-molecular bioregulators seems to be unclear. Besides its diverse functions in cells, NAD+ serves as a major source of ribofuranosyl moiety in transfer reactions and disaccharide nucleoside formation and thus plays diverse bioregulatory functions in cells. PAR, a key regulator biopolymer, is enzymatically synthesized from NAD+ and takes part in DNA repair and in a variety of metabolic pathologies associated with triggering of cell death. PAR consists of ADPR moieties joined with O-glycosidic bonds. Due to its structural complexity and lability, a detailed mechanism of PAR interactions with regulatory proteins seems to be unclear. Therefore, chemical synthesis is highly needed to obtain regular PAR oligomers of fixed length. One of the urgent tasks will be an elucidation of the role of PAR in parthanatos, a mechanism of cell death, which is associated with the cytotoxic effects of PAR and the progression of socially significant pathologies, but it is not clear, whether a mechanism of parthanatos can be applied in cancer therapy. Therefore, the design of PAR analogs based on disaccharide nucleosides stable to cleavage by PARG may lead both to PARP/PARG inhibition and trigger parthanatos mechanisms inside a cell. There is also a possibility in this way to mimic PAR action by some charged natural biopolymers, e.g., heparin, chitosan, etc. Based on PARP inhibition strategy, disaccharide nucleosides mimicking PAR core motifs can be used for elaboration on their basis drug prototypes for combined anticancer therapy. During evolution in animals, diverse molecular mechanisms have been developed associated with ADPR transfer by host PARPs (ARTs) in response to viral infection. Some viruses possess proteins with ADPR-hydrolyzing activity to block a host immune response. Therefore, a way of antiviral therapy is possible to use structural analogs ADPR as inhibitors of viral enzymes and proteins, as was demonstrated by recent publications. As ADPR is an anchor of PARylation, the role of PAR in antiviral response stays open. Disaccharide nucleoside containing compounds are also formed by intramolecular NAD+ cyclization which proceeds to afford 5',5''-dinucleotides (cADPR in animals, 2'-O-cADPR and 3'-O-cADPR in plants). These NAD+ metabolites are formed under the catalytic action of various proteins (TIR/SARM, CDs) and are known to activate/block innate immunity by affection calcium release. As the structure of 2'-O-cADPR and 3'-O-cADPR was elucidated quite recently their functions are to be further studied [96]. Besides interaction with receptors, alternate mechanisms of action can be proposed [97]. According to the biological diversity of biochemical ribosylation, novel RNA components may also exist [1]. For example, 2'-O-α-D-ribofuranosyluridine, a component of a previously unknown metabolite, hellecaucaside A, was isolated from Helleborus caucasicus [98]. It was earlier found, that RNA capping by ADPR takes place in archaea by enzymatic transfer of cADPR on 5'-end of RNA [99]. Therefore, analogous molecular mechanisms may also proceed in plants and even animals. Interestingly, 3'-O-β-D-ribofuranosyladenosine, which has an identical glycosidic linkage to 3'-cADPR, has been shown to accumulate in leaves infected and can be considered as a marker of phytopathogenic injury [100–102]. Naturally occurring cyclic 5',5''-dinucleotides and their synthetic analogs possess a therapeutic potential as immunostimulators/immunosuppressants and regulators of calcium metabolic levels. Therefore, convenient and effective chemical procedures to perform a scale synthesis of cADPR and their analogs are in demand.
ADP: adenosine diphosphate
ADPR: adenosine diphosphate ribose
Arp: 2'-O-β-D-ribofuranosyladenosine-5''-phosphate
cADPR: cyclic adenosine diphosphate ribose
cIDPR: cycloinosinediphosphoribose
Grp: 2'-O-β-D-ribofuranosylguanosine-5''-phosphate
NAD+: nicotinamide adenine dinucleotide
PAR: poly(adenosine diphosphate ribose)
PARG: poly(adenosine diphosphate ribose) glycohydrolase
PARP: poly(adenosine diphosphate ribose) polymerase
TIR: Toll/interleukin-1 receptor
tRNA: transfer RNA
tRNAMet: methionine transfer RNA
DP: Conceptualization, Formal analysis, Writing—original draft. AK: Formal analysis, Validation. CA: Writing—review & editing, Funding acquisition. MD: Conceptualization, Writing—original draft, Writing—review & editing, Funding acquisition, Supervision.
The authors declare no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
This research was funded by the Program of Fundamental Research in the Russian Federation for the 2021–2030 period “Development of biologically active analogues of natural compounds with potential use in pharmaceutics and biomedicine” [project No. 124022200001-4]. The funders had roles in data collection and analysis, but had no role in study design, decision to publish, or preparation of the manuscript.
© The Author(s) 2025.
Open Exploration maintains a neutral stance on jurisdictional claims in published institutional affiliations and maps. All opinions expressed in this article are the personal views of the author(s) and do not represent the stance of the editorial team or the publisher.
Copyright: © The Author(s) 2025. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

View: 1354
Download: 59
Times Cited: 0
Lakshmi Mounika Kelam ... M. Elizabeth Sobhia