Original Article
Original Article

Affiliation:
1U1286-INFINITE-Lille Inflammation Research International Center, ICPAL, University of Lille, Inserm, 59000 Lille, France
ORCID: https://orcid.org/0000-0001-6336-3923
Affiliation:
2UMR9020-UMR1277-Canther-Cancer Heterogeneity, Plasticity and Resistance to Therapies, University of Lille, CHU Lille, CNRS, Inserm, 59000 Lille, France
3Oncowitan, Scientific Consulting Office, 59290 Lille, France
Email: christian.bailly@univ-lille.fr
ORCID: https://orcid.org/0000-0002-2973-9357
Explor Drug Sci. 2025;3:1008113 DOl: https://doi.org/10.37349/eds.2025.1008113
Received: March 14, 2025 Accepted: May 05, 2025 Published: June 09, 2025
Academic Editor: Francisco Javier Luque Garriga, University of Barcelona, Spain
Aim: The immunosuppressive drug brequinar (BQR) is a potent inhibitor of dihydroorotate dehydrogenase (DHODH) active against autoimmune diseases and viral infections. This oral drug is currently evaluated for the treatment of cancers, notably acute myeloid leukemia to limit the suppressive function of myeloid cells. A combination of BQR and an anti-PD-1 (programmed death-1) antibody has revealed potent antitumor and antimetastatic activities. BQR induced a marked down-regulation of PD-L1 (programmed death-ligand 1) gene expression and a large decrease of PD-L1 protein expression in implanted tumors in mice.
Methods: The present study evaluated the capacity of BQR to interact directly with the PD-L1 protein dimer using molecular modeling.
Results: Molecular docking experiments revealed a modest capacity of BQR to stabilize PD-L1 dimers. The PD-L1 binding capacities of four known BQR analogs were compared to establish structure-binding relationships. The protein binding was significantly enhanced when the acid function of BQR was replaced with a trifluoroethanol substituent. The interaction was further reinforced when BQR was coupled to a mitochondria-targeted triphenylphosphine (TPP) unit. Among three BQR-TPP hybrids, compound B2 with a short alkyl linker revealed a prominent capacity to interact with PD-L1, superior to that of the reference biphenyl ligand BMS-202.
Conclusions: Two PD-L1 binders derived from BQR have been identified and the protein interaction modeled. Our study underlines the possibility of designing novel small molecule ligands targeted to the PD-L1 dimer interface based on the BQR scaffold.
Brequinar (BQR) is an immunosuppressive drug developed in the early 1990s for the prevention of rejection of organ grafts and the treatment of autoimmune diseases [1, 2]. The drug has been advanced to phase II clinical trials for the treatment of transplant rejection but trials have been discontinued, apparently because of a narrow therapeutic index. BQR showed a tendency to accumulate when given daily per os and its pharmacokinetic interaction with cyclosporine enhanced its toxicity. BQR induced leukocytopenia, thrombocytopenia, thymic atrophy, and cellular depletion of bone marrow in addition to villous atrophy in the jejunum [3, 4].
From a mechanistic viewpoint, BQR is a selective, highly potent, and slow-binding inhibitor of human dihydroorotate dehydrogenase (DHODH, IC50 = 10 nM), a ubiquitous mitochondrial enzyme essential for de novo pyrimidine biosynthesis (Figure 1) [5, 6]. As such, BQR induces a global depletion of nucleotides, both purines and pyrimidines, notably in T and B cells, and a suppression of the lymphocyte proliferative response [7].
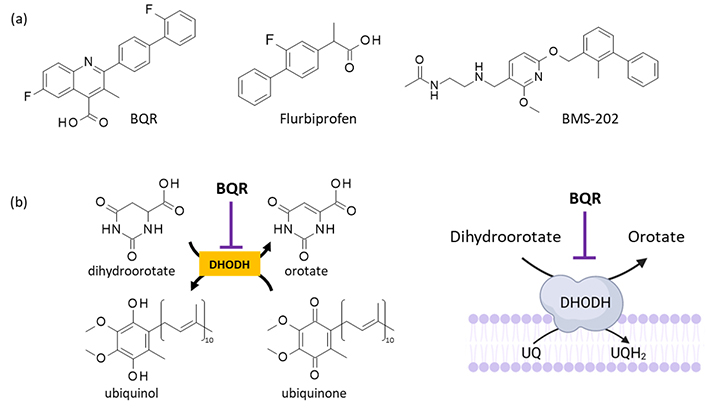
BQR and its mechanism of action. (a) Structure of BQR (NSC 368390 or DuP 785) and two PD-L1-binding compounds bearing a diphenyl unit. (b) Mechanism of action of BQR as an inhibitor of human DHODH. BQR: brequinar; DHODH: dihydroorotate dehydrogenase; PD-L1: programmed cell death-ligand 1; UQ: ubiquinone; UQH2: ubiquinol. Figure 1b is adapted with permission from [53] (© 2013 Elsevier B.V.) and [54] (© 2023 MyJoVE Corporation)
DHODH represents a therapeutic target for multiple diseases, including cancer [8]. The anticancer activity of DHODH inhibitors is linked, at least in part, to the high demand for pyrimidine nucleotides for cancer cell proliferation [9]. Inhibition of human DHODH induces a rapid depletion of intracellular uridine monophosphate (UMP) leading to the starvation of pyrimidine bases. BQR has been tested in phase II clinical trials in patients with advanced cancers of the colon, head and neck, lung, breast, and patients with gastrointestinal cancer or melanoma [10–15]. Unfortunately, BQR was found to be inactive in all types of solid tumors, possibly because of their high uridine levels [16]. Recently, the combination of BQR and dipyridamole has been re-evaluated for the treatment of neuroblastoma [17–19]. Unlike the DHODH inhibitors leflunomide and teriflunomide, BQR has not been approved for the treatment of cancer but it exhibits a robust anticancer action.
Promising results were obtained with BQR in the frame of acute myeloid leukemia (AML). BQR induces differentiation of myeloid leukemia cells via inhibition of mitochondrial complex III and induction of a pyrimidine depletion [20–23]. BQR efficiently reduces AML cell growth [24]. The capacity of BQR to limit the suppressive function of myeloid cells has prompted the development of combinations with immune checkpoint inhibitors, notably those targeting the PD-1/PD-L1 (programmed death-1/programmed death-ligand 1) checkpoint. Inhibition of DHODH with BQR has been shown to cause upregulation of antigen presentation pathway genes and cell surface MHC (major histocompatibility complex) class I expression. The combination of BQR with an immune checkpoint blocker, notably an anti-PD-1 antibody, reinforces the antitumor and antimetastatic activities of the antibody, through modulation of myeloid-derived suppressor cells (MDSC) [25, 26]. Moreover, BQR can induce a down-regulation of PD-L1 gene expression and a strong decrease of PD-L1 protein expression in cancer cells and in implanted tumors in mice. These effects concur with enhancing BQR anticancer efficacy [27].
The PD-L1 inhibitory effect of BQR can be indirect but it is certainly not a consequence of DHODH inhibition because the potent DHODH inhibitor teriflunomide has been shown to induce expression of PD-L1 and to stimulate monocytes expressing PD-L1 [28]. For this reason, we hypothesized that BQR might exert an effect on PD-L1 via a direct protein interaction. The hypothesis derives from the above-mentioned observations and from previous studies with PD-L1-binding drugs bearing a diphenyl scaffold similar to that of BQR. Indeed, numerous biphenyl-containing small molecules have been shown to bind to PD-L1 and to stabilize PD-L1 dimers, notably the reference products BMS-202 and BMS-1166 [29, 30] (Figure 1a). We have previously shown that small molecules with a biphenyl scaffold, in particular, the anti-inflammatory drug flurbiprofen (FLB) and close analogs, can form stable complexes with PD-L1 [31]. BQR contains a fluorinated biphenyl unit identical to that of FLB and the two compounds present a similar acid function (Figure 1a). Therefore, like BMS-202 and FLB, BQR could bind to PD-L1. These considerations prompted us to investigate the interaction of BQR with PD-L1 using molecular modeling. A molecular docking analysis suggested that BQR could form complexes with PD-L1 but with a reduced efficacy compared to FLB and BMS-202. For this reason, BQR analogs capable of forming more stable complexes with the checkpoint protein were searched. The in silico analysis has led to the identification of a few potent PD-L1 binders, thus opening the door to the design of a novel series of regulators of the immune checkpoint based on the BQR scaffold.
The tridimensional structure of the dimeric form of the extracellular domain of PD-L1 was retrieved from the Protein Data Bank (PDB, https://www.rcsb.org/) under the PDB code 5J89 [32]. The GOLD 5.3 software (Cambridge Crystallographic Data Centre, Cambridge, UK) was used to perform molecular docking analysis. Prior to the docking operations, the structure of each ligand was optimized using a classical Monte Carlo (MC) conformational searching procedure via the BOSS (Biochemical and Organic Simulation System) v4.9 software [33]. Molecular graphics and analysis were performed using Discovery Studio Visualizer, Biovia 2020 (Dassault Systèmes BIOVIA Discovery Studio Visualizer 2020, San Diego, Dassault Systèmes, 2020).
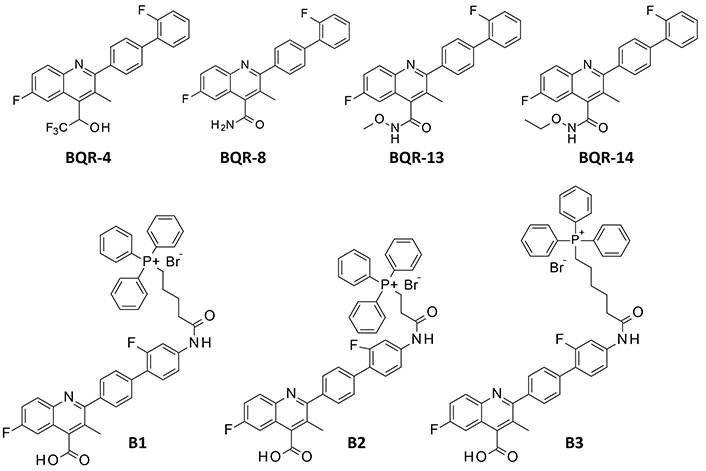
The docking analysis was performed in two steps: (i) with BQR and the reference products BMS-202 and FLB; and (ii) with BQR analogs and derivatives. Different series of BQR analogs have been designed and tested as anti-leukemic agents [34, 35]. Non-carboxylic acid derivatives of BQR have revealed an improved cellular potency compared to the parent product [36]. We selected four compounds in this series to evaluate their PD-L1 binding capacity: the fluorinated analog BQR-4, amide compound BQR-8, and the N-methoxy and N-ethoxy acetamide derivatives BQR-13 and BQR-14 (Figure 2). These four compounds have been identified as potent inhibitors of DHODH and cytotoxic agents, notably BQR-13 which inhibited DHODH more potently than the parent compound BQR and showed a higher cytotoxic potential [36].
In addition, three hybrid compounds were evaluated as potential PD-1 binders: B1, B2, and B3 (Figure 2). These molecules incorporate a BQR moiety coupled to a triphenylphosphine (TPP) to target mitochondria. They differ by the length of the (CH2)n alkyl linker between the BQR and TPP units, with n = 2, 4, or 5 for compounds B2, B1, and B3, respectively. Compound B2 has been shown to target DHODH most efficiently, triggering the formation of reactive oxygen species (ROS), promoting mitochondrial lipid peroxidation, and inducing ferroptosis in cultured cancer cells [27]. This hybrid compound showed no apparent adverse effects in mice and most importantly, it revealed an unexpected capacity to downregulate PD-L1 in cancer cells. The anticancer performance of B2 was clearly superior to that of the parent compound BRQ in a melanoma B16F10 murine model [27].
The docking procedure includes the following steps:
An MC conformational search of the ligands using the BOSS software, freely available to academic users. The best starting geometries (minimum-energy conformers) were defined for each compound. Within BOSS, MC simulations were performed in the constant-temperature and constant-pressure ensemble (NPT), as described [37].
Determination of the free energy of hydration for ligand structure. The molecular mechanics/generalized Born surface area (MM/GBSA) procedure was used to evaluate the free energies of hydration (ΔG), in relation to aqueous solubility [33]. MM/GBSA does not provide information about the number, position, and free energy of water molecules but the contribution of water molecules in the binding site is estimated in the calculation of Gibb’s free energy of binding (ΔG) values [38]. MC search and computation of ΔG were performed within BOSS using the xMCGB script as previously described [33, 39]. The best ligand structure was then considered for the docking procedure.
Definition of the protein-ligand sites of interaction. Drug-binding sites were searched using the CASTp 3.0 software for active site prediction. With the PDB structure 5J89, the flexible amino acids are Tyr56, Met115, Asp122, Tyr123, and Lys124 (monomer A), and Tyr56, Gln66, Met115, Asp122, and Tyr123 (monomer B). The side chains of these amino acids were considered fully flexible during docking. The side-chain protonation state of ionizable residues (Asp, Glu, His, Tyr, Lys) and the tautomeric forms of histidine (His) are controlled. In general, up to 100 poses considered energetically reasonable are selected during the search for the correct binding mode of the ligand. The decision to select a trial pose is based on ranked poses, using the PLP (Piecewise Linear Potential) fitness scoring function. The PLP fitness value is a pairwise additive scoring function incorporated into GOLD [40]. The same procedure was used to establish molecular models for all compounds.
Docking procedure with GOLD. A typical docking process considers 100 energetically reasonable poses (according to the ChemPLP scoring function) to search for the correct ligand binding mode. The decision to maintain a trial pose is based on ranked poses, using the PLP fitness scoring function. Six poses are kept usually. The empirical potential energy of the interaction ΔE for the ranked complexes was evaluated using the expression ΔE (interaction) = E (complex) − [E (protein) + E (ligand)]. Calculations of the final energy are performed on the basis of the SPASIBA force field. SPASIBA (integrated into CHARMM) has been specifically developed to provide refined empirical molecular mechanics force field parameters [41]. SPASIBA reproduces vibrational frequencies (with a higher accuracy than molecular mechanics potentials), and potential energy distributions of normal modes. It has been shown to be excellent at reproducing crystal-phase infrared data. The parameters are derived from vibrational wavenumbers obtained in the infrared and Raman spectra of a large series of compounds including organic molecules, amino acids, saccharides, nucleic acids, and lipids [42].
The same procedure was used to establish molecular models for all compounds. We have previously employed the same computational approach to identify PD-L1 binding small molecules, including both natural products and synthetic compounds [31, 43, 44].
BQR was docked into the cavity at the interface of two adjacent PD-L1 monomers, delimited by four tyrosine residues (two Y123 + Y56 on each side). This cavity is known to accommodate small molecules [45]. The modeling analysis indicated that BQR can form complexes with PD-L1 in a manner comparable to that observed with BMS-202. The molecule inserts its biphenyl unit at the junction of the two PD-L1 monomers, in a relatively elongated cavity. The fluoro-biphenyl portion projects deeply into the cavity in a hydrophobic region, not accessible to the solvent whereas the methylquinoline moiety is at the surface of the protein groove, as represented in Figure 3. The elongated structure of the drug is well adapted to the size of the protein cavity.
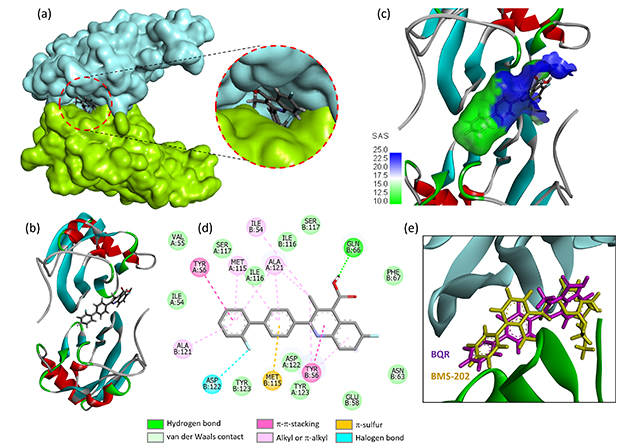
Molecular model of BQR bound to the PD-L1 dimer. Views (a) and (b) show the drug located at the interface of the protein dimer. The two protein units (in cyan and green) sandwich the small molecule ligand. The inset shows the deep insertion of the drug in the binding site. (c) A close-up view of BQR bound to the PD-L1 interface. The SAS around the drug binding zone is represented with the indicated color code. (d) Binding map contacts for BQR bound to PD-L1 (color code indicated). (e) Superimposed models of BMS-202 and BQR. BQR: brequinar; PD-L1: programmed death-ligand 1; SAS: solvent-accessible surface
BQR can form a stable complex with the PD-L1 dimer but the calculated empirical potential energy (ΔE = –59.00 kcal/mol) is significantly superior (less negative) to that calculated with the reference product BMS-202 (ΔE = –82.03 kcal/mol) under identical conditions. It is inferior to that calculated with FLB (ΔE = –47.44 kcal/mol). However, the free energy of hydration is much less favorable (ΔG = –15.02 kcal/mol for FLB vs. –5.59 kcal/mol for BQR). A number of molecular interactions, including H-bonds, π-stacking interactions, and van der Waals contacts, stabilize the drug-protein dimer complex (not shown) but the extent of binding of BQR to PD-L1 is relatively modest. Overall, the data suggest that BQR is not a robust PD-L1 binder.
We compared the capacity of compounds BQR-4, BQR-8, BQR-13, and BQR-14 to form complexes with PD-L1 using the same methodology as for BQR. From the calculated ΔE/ΔG values, some structure-binding relationships can be determined (Table 1).
Calculated potential energy of interaction (ΔE) and free energy of hydration (ΔG) for the interaction of BQR and derivatives with PD-L1
| Compounds | ΔE (kcal/mol) | ΔG (kcal/mol) |
|---|---|---|
| BMS-202 | –82.03 | –22.43 |
| Flurbiprofen | –47.44 | –15.02 |
| BQR | –59.00 | –5.59 |
| BQR-4 | –92.70 | –13.90 |
| BQR-8 | –49.85 | –6.30 |
| BQR-13 | –77.70 | –10.25 |
| BQR-14 | –80.90 | –14.50 |
| B-1 | –101.11 | –6.60 |
| B-2 | –104.10 | –13.30 |
| B-3 | –99.95 | –5.62 |
BQR: brequinar; PD-L1: programmed death-ligand 1
The replacement of the acid function of BQR with an amide (BQR-8) showed no improvement in the PD-L1 binding capacity. In contrast, the incorporation of a trifluoroethanol substituent in place of the acid group provided a derivative (BQR-4) capable of forming much more stable complexes with PD-L1 than the parent compound. The observation is interesting because this compound BQR-4 has been shown to inhibit DHODH much less efficiently than BQR (IC50 = 0.48 and 808 nM for BQR and BQR-4, respectively) while maintaining a significant cytotoxic potential [36]. BQR-4 shows a high capacity to interact with PD-L1, even superior to that of BMS-202, and this interaction may participate in compound bioactivity. Both BQR-4 and BQR-14 can form stable complexes with the PD-L1 dimer (Figure 4). Their biphenyl unit inserts at the surface of the protein dimer, while the quinoline unit remains accessible to the solvent. Different molecular contacts, notably a π-sulfur interaction with residue Met115 and π-π stacking interaction with Tyr56, stabilize the drug-protein complexes [Figure 5 with notable contacts shown in pink (π-π stacking) and in orange (π-sulfur interaction)]. Other molecular contacts, in particular, van der Waals interactions and key ligand-protein H-bonds (in green) contribute to stabilizing the molecular edifices. The multiplicity of the contacts contributes the stabilizing the drug-protein complex. The trifluoroethanol substituent of BQR-4 provides a better anchorage to the protein than the N-methoxyacetamide (BQR-13) or N-ethoxyacetamide (BQR-14) unit.
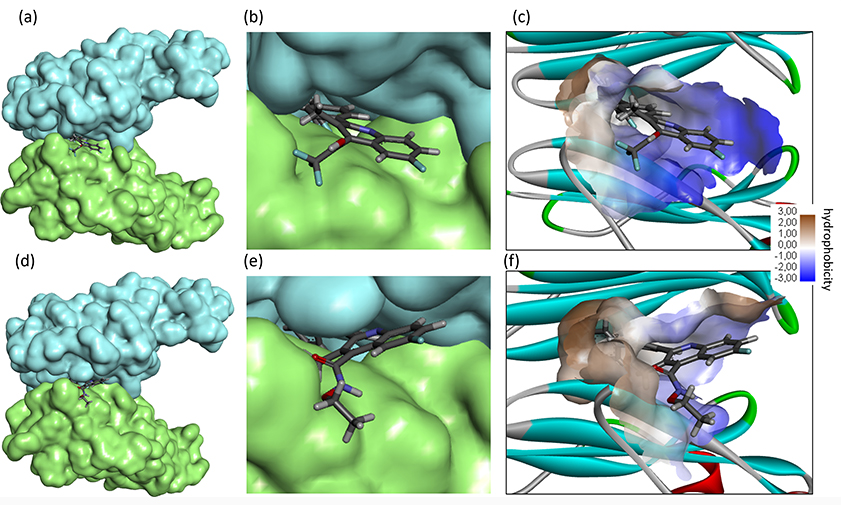
Molecular model of compounds BQR-4 (top) and BQR-14 (bottom) bound to the PD-L1 dimer. (a, d) The drug is located at the interface of the two protein units (in cyan and green). (b, e) A detailed view of the drug inserted in the binding site. (c, f) The hydrophobicity surface around the drug binding zone is represented with the indicated color code. BQR: brequinar; PD-L1: programmed death-ligand 1
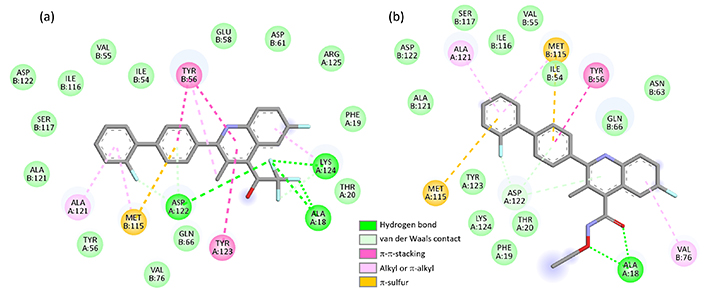
Interaction of BQR analogues with PD-L1. Binding map contacts for (a) BQR-4 and (b) BQR-14 bound to PD-L1 (color code indicated). BQR: brequinar; PD-L1: programmed death-ligand 1
The case of compound BQR-14 is interesting because this compound is a 35-fold weaker inhibitor of DHODH than compound BQR-13 (IC50 = 0.12 and 4.2 nM for BQR-13 and BQR-14, respectively) but its cytotoxic potency remains very high [36]. Apparently, its cytotoxicity is not only dependent on DHODH inhibition. Our data suggest that the BQR scaffold can be exploited to design novel PD-L1 inhibitors.
The PD-L1 binding capacity of hybrid compounds B1–B3 was compared to that of BQR. Interestingly, the docking analysis indicated that B2 can form stable complexes with PD-L1, with a PD-L1 binding capacity superior to that of analogs B1 and B3. The calculated empirical potential energy (ΔE = –104.10 kcal/mol) is favorable and the free energy of hydration (ΔG = –13.30 kcal/mol) is improved for B2 compared to B1 and B3 (Table 1). A molecular model of B2 bound to the PD-L1 dimer is shown in Figure 6. In this case, the quinoline moiety is inserted into the interfacial protein cavity maintaining the TPP unit essentially outside the cavity. Most of the molecular contacts are provided by the BQR portion of the hybrid (Figure 6d). Nevertheless, the TPP moiety participates in the stability of the drug-protein complex, through interaction with residue Tyr56. The elongated drug covers the entire protein groove, from Tyr56 on the PD-L1 unit-A to Tyr56 on the facing PD-L1 unit-B. There are over 30 drug-protein contacts ensuring good complex stability. The hybrid B2 is apparently a better PD-L1 binder than the derivative BQR-4. These observations may contribute to the reported capacity of these BQR-TPP hybrid compounds, notably the lead B2, to induce a downregulation of PD-L1 in cancer cells.
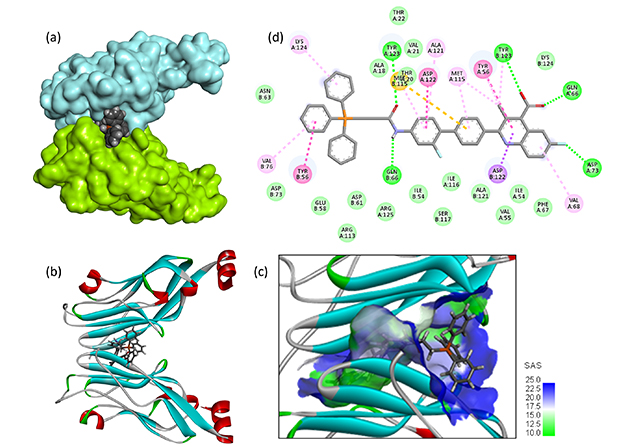
Molecular model of compound B2 bound to the PD-L1 dimer. (a, b) The drug located at the interface of the two protein units (in cyan and green). (c) The SAS around the binding site with the indicated color code. (d) Binding map contacts for B2 bound to PD-L1 (color code as in Figure 5). PD-L1: programmed death-ligand 1; SAS: solvent accessible surface
Altogether, the molecular docking analysis indicated a modest capacity of BQR to interact with PD-L1 but the extent of binding can be reinforced significantly through modulation of the drug scaffold. The replacement of the acid function on the methylquinoline unit of BQR with a trifluoroethanol substituent afforded a very interesting molecule. This compound BQR-4 is a weak DHODH inhibitor but it maintains a good capacity to inhibit cell proliferation. PD-L1 blockade may contribute partially to the bioactivity of compounds like BQR-4 and BQR-14. Similarly, the docking analysis suggested that the PD-L1 binding capacity can be enhanced upon coupling of the BQR unit with a mitochondria-targeted TPP unit. Compound B2 displays a remarkable capacity to interact with PD-L1, covering the long groove between the two PD-L1 units. The study underlines the possibility of designing novel PD-L1 binders based on the BQR scaffold.
DHODH inhibitors are under clinical study for hematologic malignancies, notably AML. BQR has demonstrated preclinical safety and good activity in murine models of AML. The drug was shown to suppress the production of MDSC from early-stage myeloid progenitors and to enhance myeloid maturation [26]. BRQ restored the terminal differentiation of MDSC [46]. Moreover, BQR was found to promote the antitumor and antimetastatic activities of an anti-PD-1 antibody in a murine model (4T1) of breast cancer, through modulation of MDSC and CD8+ T cell responses [25, 26]. The targeting of MDSC by BQR was unanticipated. In addition, liposomes loaded with BQR have been shown to facilitate the infiltration of CD8+ T cells into tumor microenvironment for enabling efficient checkpoint blockade immunotherapy [47]. In parallel, the hybrid compound B2 comprising a BQR unit coupled to a TPP unit via a short linker, induced a significant downregulation of both PD-L1 gene and PD-L1 protein expression in cancer cells and alleviated immune suppression [27]. These considerations prompted us to investigate the potential direct binding of BQR and selected derivatives to PD-L1. The fluoro-biphenyl moiety of BQR resembles that of the drug FLB which can be used to target PD-L1 expression [31, 48].
DHODH is not the unique target of BQR. The drug has been shown to selectively bind to and occupy the catalytic pocket of the oncogenic m6A RNA demethylase FTO (fat mass and obesity-associated protein) implicated in leukemia. BQR exerts its anti-leukemic effect by suppression of FTO activity and activation of apoptosis signaling [49]. DHODH and FTO, the two target enzymes of BRQ, present structural similarities at the level of their catalytic pockets enabling the simultaneous targeting of the two proteins. This dual targeting explains why BQR exhibits FTO-independent antileukemic effects [24]. In addition, a third potential activity, at the level of PD-L1 warrants further investigation. BQR itself does not emerge as a strong PD-L1 binder but the drug can be used as a template to design better binders like compound BQR-4 and B2. Our study opens novel perspectives to the design of biphenyl-containing PD-L1 binding compounds based on BQR. There exists a large set of BQR derivatives equipped with a biphenyl unit [36, 46, 50, 51]. Their PD-L1 binding capacity warrants further investigation.
The in silico study faces important limitations, inherent to the molecular docking procedure (e.g., lack of solvent dynamics or entropy considerations). The performance of a docking method for correctly predicting the binding poses of a ligand can vary significantly from one program to another, but the GOLD docking algorithm is considered a robust tool [52]. All docking methods have their specific limitations and the technical impediments shall not prevent the pursuit of novel inhibitors. There is a need for supportive studies (statistical analysis, molecular dynamics) to support the results. Experimental validation of the results will be needed also to confirm the capacity of the compound to bind to PD-L1 and to interfere with the functioning of the checkpoint. However, the in silico analysis is useful to assist in drug discovery tasks. The docking analysis facilitates a comprehensive understanding of the interaction of BQR-type compounds with the protein binding site. It is an aid in drug design and for prioritization of new drug synthesis. The study provides prospects for future researches, which need to investigate not only drug binding to PD-L1 but also their capacity to modulate PD-L1 expression in cells.
In conclusion, the molecular docking analysis suggests that BQR presents a modest capacity to bind to PD-L1 but the interaction of the drug with the checkpoint protein can be significantly enhanced upon replacement of its acid group with a trifluoroethanol substituent as in compound BQR-4. Similarly, the covalent linkage of BQR to a mitochondria-targeted TPP unit via a short alkyl linker reinforces significant drug binding to PD-L1 as in compound B2. The study opens drug design perspectives. Novel PD-L1-binding molecules incorporating a BQR-like scaffold can be designed and tested as antitumor agents. Our work introduces a novel hypothesis, with a possible contribution of PD-L1 to the mechanism of action of BQR. A novel direction is proposed for the design of innovative anticancer agents with immunomodulatory properties. However experimental validation is imperative to validate the in silico predictions.
AML: acute myeloid leukemia
BOSS: Biochemical and Organic Simulation System
BQR: brequinar
DHODH: dihydroorotate dehydrogenase
FLB: flurbiprofen
MC: Monte Carlo
MDSC: myeloid-derived suppressor cells
MM/GBSA: molecular mechanics/generalized Born surface area
PD-1: programmed death-1
PD-L1: programmed death-ligand 1
PDB: Protein Data Bank
PLP: Piecewise Linear Potential
TPP: triphenylphosphine
GV: Investigation, Visualization, Software, Methodology. CB: Conceptualization, Investigation, Writing—original draft, Writing—review & editing, Supervision. Both authors read and approved the submitted version.
Both authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
The raw data supporting the conclusions of this manuscript will be made available by the authors, without undue reservation, to any qualified researcher.
Not applicable.
© The Author(s) 2025.
Open Exploration maintains a neutral stance on jurisdictional claims in published institutional affiliations and maps. All opinions expressed in this article are the personal views of the author(s) and do not represent the stance of the editorial team or the publisher.
Copyright: © The Author(s) 2025. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

View: 2123
Download: 65
Times Cited: 0